"Cromatografía líquida de alta resolución de contaminantes naturales y de aguas residuales"
Introducción
Capítulo 1. Conceptos básicos y clasificación de los métodos de cromatografía líquida
1.1 Aparatos para cromatografía líquida
Capítulo 2. La esencia de HPLC
2.1 Aplicación
Capítulo 3. Ejemplos de uso de HPLC en el análisis de objetos ambientales
Capítulo 4 Instrumentación de HPLC
Literatura
Apéndice
Introducción
Métodos cromatográficos son a menudo indispensables para la identificación y cuantificación de compuestos orgánicos con una estructura similar. Los más utilizados para el análisis rutinario de contaminantes ambientales son la cromatografía de gases y la cromatografía líquida de alta resolución. El análisis por cromatografía de gases de contaminantes orgánicos en aguas potables y residuales se basó inicialmente en el uso de columnas empaquetadas, más tarde también se generalizaron las columnas capilares de cuarzo. El diámetro interno de las columnas capilares suele ser de 0,20-0,75 mm, longitud - 30-105 m Los resultados óptimos en el análisis de contaminantes en el agua se logran con mayor frecuencia cuando se utilizan columnas capilares con diferentes espesores de película hechas de siliconas de metilfenilo con un contenido de fenilo. grupos de 5 y 50% . El sistema de inyección de muestras a menudo se convierte en un punto vulnerable en las técnicas cromatográficas que utilizan columnas capilares. Los sistemas de inyección de muestras se pueden dividir en dos grupos: universales y selectivos. Los universales incluyen sistemas de inyección con y sin desdoblamiento del caudal, inyección “fría” en columna y evaporación con programación de temperatura. La inyección selectiva utiliza purga con captura intermedia, análisis de espacio de cabeza, etc. Cuando se utilizan sistemas de inyección universal, toda la muestra ingresa a la columna, con inyección selectiva, solo se introduce una fracción determinada. Los resultados obtenidos con la inyección selectiva son significativamente más precisos, ya que la fracción que ingresa a la columna contiene solo sustancias volátiles, y la técnica puede automatizarse por completo.
Los detectores cromatográficos de gases utilizados en el control de contaminantes se dividen a menudo en detectores universales, que responden a cada componente de la fase móvil, y detectores selectivos, que reaccionan ante la presencia en la fase móvil de un determinado grupo de sustancias con características químicas similares. Los universales incluyen ionización de llama, emisión atómica, detectores espectrométricos de masas y espectrometría infrarroja. Los detectores selectivos utilizados en el análisis del agua son de captura de electrones (selectivo para sustancias que contienen átomos de halógeno), termoiónico (selectivo para compuestos que contienen nitrógeno y fósforo), fotoionización (selectivo para hidrocarburos aromáticos), detector de conductividad electrolítica (selectivo para compuestos que contienen halógeno). , átomos de azufre y nitrógeno). Las cantidades mínimas detectables de sustancias oscilan entre nanogramos y picogramos por segundo.
Cromatografía líquida de alta resolución(HPLC) es un método ideal para la determinación de un gran número de compuestos térmicamente inestables que no pueden analizarse mediante cromatografía de gases. Actualmente, los agroquímicos modernos, incluidos los carbonatos de metilo y los insecticidas organofosforados, y otras sustancias no volátiles, a menudo se convierten en objetos de análisis por cromatografía líquida. La cromatografía líquida de alta resolución (HPLC) está ganando popularidad entre otros métodos utilizados en el monitoreo ambiental, también porque tiene grandes perspectivas en términos de automatización de la preparación de muestras.
CAPÍTULO 1. CONCEPTOS BÁSICOS Y CLASIFICACIÓN DE LOS MÉTODOS DE CROMATOGRAFÍA DE LÍQUIDOS
La cromatografía líquida se divide en varias clases según el tipo de soporte de fase estacionaria. La instrumentación simple del papel y la cromatografía en capa fina condujo al uso generalizado de estos métodos en la práctica analítica. Sin embargo, las grandes posibilidades de la cromatografía líquida en columna estimularon la mejora de los equipos para este método clásico y condujeron a la rápida introducción de la HPLC. Pasar el eluyente a través de la columna a alta presión hizo posible aumentar considerablemente la tasa de análisis y aumentar significativamente la eficiencia de separación debido al uso de un adsorbente finamente disperso. El método HPLC actualmente permite aislar, analizar cuantitativa y cualitativamente mezclas complejas de compuestos orgánicos.
Según el mecanismo de interacción de la sustancia separada (eluido) con la fase estacionaria, se distinguen la adsorción, la distribución, el intercambio iónico, la exclusión por tamaño, el par iónico, el intercambio de ligando y la cromatografía de afinidad.
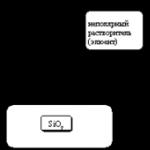
Cromatografía de adsorción. La separación por cromatografía de adsorción se lleva a cabo como resultado de la interacción de la sustancia a separar con un adsorbente, como el óxido de aluminio o el gel de sílice, que tienen centros polares activos en la superficie. El solvente (eluyente) es un líquido no polar. El mecanismo de sorción consiste en una interacción específica entre la superficie polar del adsorbente y las regiones polares (o susceptibles de ser polarizadas) de las moléculas del componente analizado (Fig. 1).
Arroz. 1. Cromatografía líquida de adsorción.
cromatografía de partición. En la versión distributiva de la cromatografía líquida, la separación de una mezcla de sustancias se lleva a cabo debido a la diferencia en sus coeficientes de distribución entre dos fases inmiscibles: el eluyente (fase móvil) y la fase ubicada en el adsorbente (fase estacionaria).
En fase normal La cromatografía líquida de partición utiliza un eluyente no polar y grupos polares injertados en la superficie de un adsorbente (generalmente gel de sílice). Los alquilclorosilanos sustituidos que contienen grupos polares, como nitrilo, grupo amino, etc., se utilizan como modificadores de la superficie del gel de sílice (fases unidas) (Fig. 2). El uso de fases ligadas permite controlar con precisión las propiedades de sorción de la superficie de la fase estacionaria y lograr una alta eficiencia de separación.

Arroz. 2. Cromatografía de partición con fase ligada (variante de fase normal).
fase inversa la cromatografía líquida se basa en la distribución de los componentes de la mezcla entre el eluyente polar y los grupos no polares (cadenas alquílicas largas) injertados en la superficie del sorbente (Fig. 3).

Arroz. 3. Cromatografía de partición con fase ligada (versión de fase reversa).
Menos utilizada es una variante de la cromatografía líquida soportada, en la que se aplica una fase estacionaria líquida a un soporte estacionario.
Exclusivo (gel penetrante) La cromatografía es una variante de la cromatografía líquida en la que la separación de sustancias ocurre debido a la distribución de moléculas entre el solvente ubicado en los poros del adsorbente y el solvente que fluye entre sus partículas.
afín La cromatografía se basa en interacciones específicas de proteínas separadas (anticuerpos) con sustancias (antígenos) injertadas en la superficie de un sorbente (resina sintética) que forma selectivamente complejos (conjugados) con proteínas.
La cromatografía de intercambio de iones, de pares de iones y de intercambio de ligandos se utiliza principalmente en el análisis inorgánico.
Parámetros básicos de la separación cromatográfica.
Los principales parámetros de la separación cromatográfica son el volumen de retención y el tiempo de retención del componente de la mezcla (Fig. 4).
El tiempo de retención tR es el tiempo transcurrido desde que se inyecta la muestra en la columna hasta que se alcanza el máximo del pico correspondiente. Multiplicando el tiempo de retención por la velocidad del volumen de eluyente F, obtenemos el volumen de retención VR:
El tiempo de retención corregido es el tiempo transcurrido desde que aparece el pico del componente no absorbible hasta el pico del compuesto correspondiente:
tR" = tR - t0 ;
El volumen de retención normalizado o corregido es el volumen de retención corregido por el volumen muerto de la columna V0, es decir, el volumen de retención del componente no absorbible:
VR" = VR - V0;
La característica de retención es también el factor de capacitancia k", definido como la relación entre la masa de la sustancia en la fase estacionaria y la masa de la sustancia en la fase móvil: k" = mn / mp;
El valor de k" es fácil de determinar a partir del cromatograma:

Los parámetros más importantes de la separación cromatográfica son su eficiencia y selectividad.
La eficiencia de la columna, medida por la altura de los platos teóricos (HETP) e inversamente proporcional a su número (N), es mayor cuanto más estrecho es el pico de la sustancia que emerge en el mismo tiempo de retención. El valor de eficiencia se puede calcular a partir del cromatograma utilizando la siguiente fórmula:
N = 5,54. (tR / 1/2) 2 ,
donde tr- tiempo de espera,
w 1/2 - ancho de pico a media altura
Conociendo el número de platos teóricos por columna, la longitud de la columna L y el diámetro de grano promedio dc del sorbente, es fácil obtener los valores de la altura equivalente del plato teórico (HETP) y la altura reducida (PHETP):
HETP = L/N PHETP = HETP/d c
Estas características permiten comparar la eficiencia de columnas de diferentes tipos, evaluar la calidad del adsorbente y la calidad del llenado de las columnas.
La selectividad de la separación de dos sustancias está determinada por la ecuación:
Al considerar la separación de una mezcla de dos componentes, el grado de separación RS también es un parámetro importante:
 ;
;
Los picos se consideran resueltos si el valor de RS es mayor o igual a 1,5.
Los principales parámetros cromatográficos relacionan la siguiente ecuación para la resolución:
 ;
;
Los factores que determinan la selectividad de separación son:
1) la naturaleza química del adsorbente;
2) la composición del solvente y sus modificadores;
3) estructura química y propiedades de los componentes de la mezcla a separar;
4) temperatura de la columna
1.1 Aparatos para cromatografía líquida
En la cromatografía líquida moderna, se utilizan instrumentos de diversos grados de complejidad, desde los sistemas más simples hasta cromatógrafos de alta gama equipados con varios dispositivos adicionales.
En la fig. La Figura 4 muestra un diagrama de bloques de un cromatógrafo de líquidos que contiene el conjunto mínimo requerido de componentes, de una forma u otra, presentes en cualquier sistema cromatográfico.

Arroz. 4. Diagrama de bloques de un cromatógrafo de líquidos.
La bomba (2) está diseñada para crear un flujo de disolvente constante. Su diseño está determinado principalmente por la presión de operación en el sistema. Para operar en el rango de 10-500 MPa, se utilizan bombas de émbolo (jeringa) o de pistón. La desventaja de la primera es la necesidad de paradas periódicas para el llenado con eluyente, y la segunda es la gran complejidad del diseño y, en consecuencia, el alto precio. Para sistemas simples con presiones de funcionamiento bajas de 1-5 MPa, se utilizan con éxito bombas peristálticas económicas, pero dado que es difícil lograr una presión y un caudal constantes, su uso se limita a tareas preparatorias.
El inyector (3) asegura que se inyecta en la columna una muestra de la mezcla de componentes separados con una reproducibilidad suficientemente alta. Los sistemas simples de inyección de muestra de "flujo detenido" requieren que la bomba se detenga y, por lo tanto, son menos convenientes que las pipetas de bucle de Reodyne.
Las columnas de HPLC (4) son tubos de acero inoxidable de paredes gruesas capaces de soportar altas presiones. La densidad y la uniformidad del relleno de la columna con un sorbente juegan un papel importante. Para la cromatografía de líquidos a baja presión, se utilizan con éxito columnas de vidrio de paredes gruesas. La constancia de la temperatura está asegurada por el termostato (5).
Los detectores (6) para cromatografía líquida tienen una celda de flujo en la que se mide continuamente alguna propiedad del eluyente que fluye. Los tipos más populares de detectores de propósito general son los refractómetros, que miden el índice de refracción, y los detectores espectrofotométricos, que miden la absorbancia de un solvente a una longitud de onda fija (generalmente en la región ultravioleta). Las ventajas de los refractómetros (y las desventajas de los espectrofotómetros) incluyen una baja sensibilidad al tipo de compuesto que se determina, que puede no contener grupos cromóforos. Por otro lado, el uso de refractómetros está limitado a sistemas isocráticos (con composición de eluyente constante), por lo que el uso de un gradiente de disolvente no es posible en este caso.
Las columnas de HPLC, que se utilizan con mayor frecuencia en el análisis de contaminantes ambientales, tienen una longitud de 25 cm y un diámetro interior de 4,6 mm y están llenas de partículas de gel de sílice esféricas de 5-10 µm injertadas con grupos octadecil. En los últimos años han aparecido columnas con diámetros internos más pequeños llenas de partículas más pequeñas. El uso de tales columnas reduce el consumo de solventes y la duración del análisis, aumenta la sensibilidad y la eficiencia de separación, y también facilita el problema de conectar las columnas a los detectores espectrales. Las columnas con un diámetro interno de 3,1 mm están equipadas con un cartucho de seguridad (precolumna) para aumentar la vida útil y mejorar la reproducibilidad de los análisis.
Como detectores en los instrumentos modernos de HPLC, generalmente se utilizan un detector UV en una matriz de diodos, fluorescencia y detectores electroquímicos.
Debe tenerse en cuenta que en el trabajo práctico, la separación a menudo se produce no por uno, sino por varios mecanismos simultáneamente. Por lo tanto, la separación por exclusión puede complicarse por los efectos de adsorción, adsorción - distribución y viceversa. Al mismo tiempo, cuanto mayor sea la diferencia en las sustancias de la muestra en términos del grado de ionización, basicidad o acidez, en términos de peso molecular, polarizabilidad y otros parámetros, mayor será la probabilidad de un mecanismo de separación diferente para tales sustancias
En la práctica, la cromatografía de "fase reversa" (partición), en la que la fase estacionaria no es polar, pero la fase móvil es polar (es decir, el reverso de la cromatografía de "fase directa"), se ha generalizado más.
En la mayoría de los laboratorios del mundo, se analiza un grupo de 16 HAP prioritarios mediante HPLC o CMS.
CAPÍTULO 2. ESENCIA DE LA HPLC
En la cromatografía líquida de alta resolución (HPLC), la naturaleza de los procesos que ocurren en la columna cromatográfica es generalmente idéntica a los procesos en la cromatografía de gases. La diferencia está solo en el uso de un líquido como fase estacionaria. Debido a la alta densidad de las fases móviles líquidas y la alta resistencia de las columnas, la cromatografía de gases y líquidos difiere mucho en la instrumentación.
En la HPLC se suelen utilizar disolventes puros o sus mezclas como fases móviles.
Para crear una corriente de disolvente puro (o mezclas de disolventes), denominada eluyente en cromatografía líquida, se utilizan bombas, que forman parte del sistema hidráulico del cromatógrafo.
La cromatografía de adsorción se lleva a cabo como resultado de la interacción de una sustancia con adsorbentes, como gel de sílice u óxido de aluminio, que tienen centros activos en la superficie. La diferencia en la capacidad de interactuar con los centros de adsorción de diferentes moléculas de muestra conduce a su separación en zonas en el proceso de movimiento con la fase móvil a través de la columna. La separación de las zonas de los componentes lograda en este caso depende de la interacción tanto con el solvente como con el adsorbente.
Los adsorbentes de gel de sílice con diferentes volúmenes, superficies y diámetros de poro encuentran la mayor aplicación en HPLC. El óxido de aluminio y otros adsorbentes se usan con mucha menos frecuencia. La razón principal de esto:
Resistencia mecánica insuficiente, que no permite el envasado y uso a presiones elevadas típicas de HPLC;
el gel de sílice en comparación con el óxido de aluminio tiene un rango más amplio de porosidad, superficie y diámetro de poro; una actividad catalítica significativamente mayor del óxido de aluminio conduce a una distorsión de los resultados del análisis debido a la descomposición de los componentes de la muestra o su quimisorción irreversible.
detectores de HPLC
La cromatografía líquida de alta resolución (HPLC) se utiliza para detectar sustancias polares no volátiles que, por alguna razón, no se pueden convertir en una forma conveniente para la cromatografía de gases, incluso en forma de derivados. Tales sustancias, en particular, incluyen ácidos sulfónicos, tintes solubles en agua y algunos pesticidas, como derivados de fenilurea.
Detectores:
UV - detector de matriz de diodos. La "matriz" de fotodiodos (hay más de doscientos) registra constantemente señales en la región UV y visible del espectro, lo que garantiza el registro de espectros UV-B en el modo de escaneo. Esto hace posible registrar continuamente, a alta sensibilidad, espectros sin distorsiones de componentes que pasan rápidamente a través de una celda especial.
En comparación con la detección de longitud de onda única, que no proporciona información sobre la "pureza" del pico, la capacidad de comparar los espectros completos de la matriz de diodos proporciona un resultado de identificación con un grado de certeza mucho mayor.
Detector fluorescente. La gran popularidad de los detectores fluorescentes se debe a su muy alta selectividad y sensibilidad, y al hecho de que muchos contaminantes ambientales emiten fluorescencia (por ejemplo, los hidrocarburos poliaromáticos).
Un detector electroquímico se utiliza para detectar sustancias que se oxidan o reducen fácilmente: fenoles, mercaptanos, aminas, derivados nitro aromáticos y halógenos, aldehídos, cetonas, bencidinas.
La separación cromatográfica de la mezcla en la columna debido al lento avance del PF lleva mucho tiempo. Para acelerar el proceso, la cromatografía se realiza bajo presión. Este método se denomina cromatografía líquida de alta resolución (HPLC).
La modernización del equipo utilizado en la cromatografía en columna líquida clásica lo ha convertido en uno de los métodos de análisis más prometedores y modernos. La cromatografía líquida de alta resolución es un método conveniente para separar, aislar preparativamente y realizar análisis cualitativos y cuantitativos de compuestos termolábiles no volátiles de bajo y alto peso molecular.
Según el tipo de sorbente utilizado en este método, se utilizan 2 variantes de cromatografía: en un sorbente polar que utiliza un eluyente no polar (opción de fase directa) y en un sorbente no polar que utiliza un eluyente polar, el llamado inverso cromatografía líquida de alta resolución (RPHLC) de dos fases.
Cuando el eluyente pasa al eluyente, el equilibrio en condiciones de RPHLC se establece muchas veces más rápido que en las condiciones de adsorbentes polares y PF no acuosos. Como resultado de esto, además de la conveniencia de trabajar con agua y eluyentes hidroalcohólicos, RPHLC ahora ha ganado gran popularidad. La mayoría de los análisis de HPLC se llevan a cabo utilizando este método.
detectores El registro de la salida de la columna de un componente separado se realiza mediante un detector. Para el registro, puede utilizar el cambio en cualquier señal analítica proveniente de la fase móvil y relacionada con la naturaleza y cantidad del componente de la mezcla. La cromatografía líquida utiliza señales analíticas como la absorción de luz o la emisión de luz de la solución que sale (detectores fotométricos y fluorimétricos), índice de refracción (detectores refractométricos), conductividad eléctrica y potencial (detectores electroquímicos), etc.
La señal detectada continuamente es registrada por el registrador. El cromatograma es una secuencia de señales del detector registradas en la cinta de registro, generada cuando los componentes individuales de la mezcla salen de la columna. En el caso de separación de la mezcla, los picos individuales son visibles en el cromatograma externo. La posición del pico en el cromatograma se utiliza con fines de identificación de la sustancia, la altura o el área del pico, con fines de determinación cuantitativa.
2.1 Aplicación
HPLC encuentra la aplicación más amplia en las siguientes áreas de análisis químico (se identifican objetos de análisis donde HPLC prácticamente no tiene competencia):
· Control de calidad de los alimentos: aditivos tónicos y de sabor, aldehídos, cetonas, vitaminas, azúcares, colorantes, conservantes, hormonas, antibióticos, triazina, carbamato y otros pesticidas, micotoxinas, nitrosoaminas, hidrocarburos aromáticos policíclicos, etc.
· Protección del medio ambiente: fenoles, nitrocompuestos orgánicos, hidrocarburos aromáticos monocíclicos y policíclicos, varios pesticidas, aniones y cationes principales.
· Criminalística - drogas, explosivos y colorantes orgánicos, productos farmacéuticos potentes.
· Industria farmacéutica - hormonas esteroides, prácticamente todos los productos de síntesis orgánica, antibióticos, preparados poliméricos, vitaminas, preparados proteicos.
Medicina: las sustancias bioquímicas y medicinales enumeradas y sus metabolitos en fluidos biológicos (aminoácidos, purinas y pirimidinas, hormonas esteroides, lípidos) en el diagnóstico de enfermedades, determinando la tasa de excreción de drogas del cuerpo con el propósito de su dosificación individual .
· Agricultura - determinación de nitratos y fosfatos en suelos para determinar la cantidad necesaria de fertilizantes, determinación del valor nutritivo de los piensos (aminoácidos y vitaminas), análisis de plaguicidas en suelos, aguas y productos agrícolas.
Bioquímica, química bioorgánica, ingeniería genética, biotecnología - azúcares, lípidos, esteroides, proteínas, aminoácidos, nucleósidos y sus derivados, vitaminas, péptidos, oligonucleótidos, porfirinas, etc.
· Química orgánica - todos los productos estables de síntesis orgánica, colorantes, compuestos termolábiles, compuestos no volátiles; química inorgánica (prácticamente todos los compuestos solubles en forma de iones y compuestos complejos).
· Control de calidad y seguridad de productos alimenticios, bebidas alcohólicas y no alcohólicas, agua potable, productos químicos domésticos, perfumes en todas las etapas de su producción;
determinación de la naturaleza de la contaminación en el lugar de un desastre o emergencia provocado por el hombre;
detección y análisis de sustancias estupefacientes, potentes, venenosas y explosivas;
determinación de la presencia de sustancias nocivas (hidrocarburos policíclicos y otros aromáticos, fenoles, pesticidas, colorantes orgánicos, iones de metales pesados, alcalinos y alcalinotérreos) en efluentes líquidos, emisiones al aire y desechos sólidos de empresas y en organismos vivos;
· seguimiento de procesos de síntesis orgánica, procesamiento de petróleo y carbón, producciones bioquímicas y microbiológicas;
análisis de la calidad del suelo para la fertilización, la presencia de pesticidas y herbicidas en el suelo, el agua y los productos, así como el valor nutricional de los alimentos; tareas analíticas de investigación complejas; obteniendo una microcantidad de sustancia ultrapura.
CAPÍTULO 3. EJEMPLOS DE USO DE HPLC EN EL ANÁLISIS DE OBJETOS AMBIENTALES
HPLC: un método para monitorear PAH en objetos ambientales
Para los hidrocarburos aromáticos policíclicos (PAH), ecotóxicos de la primera clase de peligro, se han establecido niveles extremadamente bajos de concentraciones máximas permisibles (MAC) en objetos naturales. La determinación de PAH al nivel de MPC e inferior es una de las tareas analíticas más complejas y se utilizan métodos de análisis de alta tecnología (GC-MS, GC, HPLC) para resolverlas. Al elegir un método para monitorear, además de las características principales en consideración, se agregan sensibilidad y selectividad, expresividad y economía, porque. el seguimiento implica un análisis en serie. La variante de HPLC en columnas cortas de pequeño diámetro cumple en gran medida estos requisitos. Utilizando este método, los autores desarrollaron y certificaron métodos para monitorear el benzo[a]pireno en tres medios naturales: aerosoles, cubierta de nieve y aguas superficiales. Los métodos se caracterizan por: preparación simple de muestras unificadas, incluida la extracción de PAH con disolventes orgánicos y concentración del extracto, introducción directa de un extracto concentrado en una columna cromatográfica, uso de detección fotométrica de múltiples longitudes de onda en la región UV del espectro, identificación de picos de PAH en cromatogramas utilizando dos parámetros, tiempo de retención y relación espectral. El error total no supera el 10 % al determinar benz[a]pireno en aerosol en el rango de concentración de 0,3 a 450 ng/m hasta 50 μg/m 2 . Para el caso de determinación simultánea de HAP prioritarios (hasta 12 compuestos) y registro de picos no homogéneos de analitos, se propuso reseparar el extracto con un cambio en la selectividad de la fase móvil, la longitud de onda de detección y la temperatura de la columna, teniendo en cuenta las propiedades individuales de los PAH que se están determinando.
1 . Calidad del aire ambiente. Concentración en masa de benzo[a]pireno. El procedimiento para realizar mediciones por el método HPLC. Certificado de atestación MVI No. 01-2000.
2 . La calidad de las aguas residuales superficiales y tratadas. Concentración en masa de benzo[a]pireno. El procedimiento para realizar mediciones por el método HPLC. Certificado de constancia MVI No. 01-2001.
3 . Calidad de la capa de nieve. Concentración en masa de benzo[a]pireno. El procedimiento para realizar mediciones por el método HPLC. Certificado de constancia MVI No. 02-2001.
Eliminación de anilina de soluciones acuosas utilizando desechos de recuperación aluminotérmica de cascarilla de cobre laminado
El problema de la remoción de hidrocarburos de las aguas residuales es una tarea urgente. En muchas industrias químicas, petroquímicas y otras, se forma anilina y sus derivados, que son sustancias tóxicas. La anilina es una sustancia altamente tóxica, MPC - 0,1 mg / m 3. La anilina y sus derivados son solubles en agua y, por lo tanto, no pueden eliminarse por sedimentación gravitacional.
Uno de los mejores métodos de tratamiento de aguas residuales de contaminantes orgánicos es el uso de adsorbentes inorgánicos y orgánicos con capacidad de regeneración (aluminosilicatos, arcillas modificadas, madera, fibras, etc.) e incapaces de regeneración (carbón activado, materiales poliméricos macroporosos, etc.) . ).
Los adsorbentes regenerados pueden eliminar sustancias orgánicas de diferente polaridad del agua. La búsqueda de adsorbentes efectivos es una tarea urgente.
Este informe presenta los resultados de un estudio en el campo del uso de la cascarilla de cobre molido de la planta de cable de Ereván (OPMOERKZ) como adsorbentes de anilina.
Los estudios cromatográficos se realizaron en un cromatógrafo HPLC/cromatografía líquida de alta resolución/sistemas (Waters 486 - detector, Waters 600S - controlador, Waters 626 - Bomba), en una columna de 250 x 4 mm llena de sorbentes en estudio, fase móvil tasa 1 ml/m/fase móvil son los solventes que estamos estudiando/, el detector es UV-254. El análisis espectroscópico UV se realizó en un espectrofotómetro Specord-50, los espectros se obtuvieron utilizando el programa informático ASPECT PLUS.
Se añadieron porciones de adsorbentes pesados con precisión a ciertos volúmenes de anilina en agua, cuyas concentraciones iniciales variaron. La mezcla se agitó intensamente durante 6 horas y luego se dejó reposar la muestra. La adsorción se completa en casi 48 horas.La cantidad de anilina precipitada se determinó por espectrofotometría UV así como por análisis refractivo.
En un primer momento, se estudiaron las propiedades de adsorción de OPMOEPKZ durante la eliminación de anilina de una solución en tetracloruro de carbono. Resultó que la anilina absorbe mejor el sorbente 3 (tabla).
También se realizaron mediciones para soluciones acuosas de anilina a concentraciones de 0,01-0,0001 mol/l. La tabla muestra los datos de una solución de 0,01 M.
Absorción de anilina por varios sorbentes de una solución acuosa de anilina 0,01 M a 20 °C
Previamente se encontró que la adsorción dentro de los rangos de concentración indicados aumenta y depende linealmente del índice de refracción. La cantidad de anilina se determinó a partir del gráfico del índice de refracción frente a la concentración molar y se corrigió tanto por cromatografía líquida como por análisis espectral UV.
El sorbente 3 es el más activo para soluciones acuosas.La cantidad de contaminante adsorbido se calculó como la diferencia entre la cantidad total de contaminante añadida a la solución inicial y su residuo en la solución final.
Métodos para determinar HAP en objetos ambientales
Por lo general, los métodos de cromatografía de gases (GC) y cromatografía líquida de alta resolución (HPLC) se utilizan para determinar los PAH. la separación de los 16 HAP principales, suficiente para el análisis cuantitativo, se logra utilizando columnas capilares en cromatografía de gases o columnas de alto rendimiento utilizadas en HPLC. Debe recordarse que una columna que separa bien las mezclas de calibración de dieciséis PAH no garantiza que también estarán bien separadas en el contexto de los compuestos orgánicos acompañantes en las muestras en estudio.
Para simplificar el análisis, así como para lograr una alta calidad de los resultados obtenidos, la mayoría de los procedimientos analíticos contienen la etapa de aislamiento preliminar (separación) de los PAH de otros grupos de compuestos relacionados en las muestras. Los métodos más comunes utilizados para este fin son la cromatografía líquida de baja presión en sistemas líquido-sólido o líquido-líquido mediante mecanismos de adsorción, como el uso de gel de sílice o alúmina, en ocasiones se utilizan mecanismos mixtos, como la adsorción y la exclusión mediante Sephadex.
El uso de pretratamiento de muestras permite evitar la influencia de:
Compuestos completamente no polares como hidrocarburos alifáticos;
Compuestos moderada y fuertemente polares, por ejemplo, ftalano, fenoles, alcoholes polihídricos, ácidos;
Compuestos de alto peso molecular como, por ejemplo, resinas.
En la cromatografía líquida de alta resolución (HPLC) se utilizan principalmente dos tipos de detectores: un detector fluorimétrico o un detector espectrofotométrico de barra de fotodiodo. El límite de detección de PAH en la detección fluorimétrica es muy bajo, lo que hace que este método sea especialmente adecuado para la determinación de cantidades traza de compuestos poliaromáticos. Sin embargo, los detectores fluorimétricos clásicos prácticamente no proporcionan información sobre la estructura del compuesto en estudio. Los diseños modernos permiten registrar espectros de fluorescencia que son característicos de compuestos individuales, pero aún no se han generalizado en la práctica de las mediciones de rutina. Un detector espectrofotométrico con línea de fotodiodo (PDL) permite registrar espectros de absorción en el rango espectral UV y visible, estos espectros se pueden utilizar para la identificación. Se puede obtener información similar usando detectores de escaneo rápido.
A la hora de elegir una técnica analítica para la separación, identificación y cuantificación de estos HAP, se deben tener en cuenta las siguientes condiciones:
El nivel de contenidos determinados en las muestras estudiadas;
El número de sustancias relacionadas;
Procedimiento analítico aplicado (procedimiento de medición);
Las posibilidades del equipo de serie.
Desarrollo de un método para la determinación de elementos alcalinotérreos y magnesio por cromatografía líquida iónica de alta resolución
El desarrollo y mejora de métodos que permitan resolver problemas de análisis de aguas es un problema importante en la química analítica. El desarrollo de la cromatografía líquida de alta presión y alto rendimiento estimuló el desarrollo de una nueva dirección en la cromatografía de intercambio iónico, la denominada cromatografía iónica. La síntesis de adsorbentes para cromatografía iónica es difícil, ya que se les imponen muchos requisitos. Debido a la falta de intercambiadores de cationes de alto rendimiento comercialmente disponibles, se utilizó una fase inversa modificada dinámicamente, para lo cual se sintetizó un modificador: ácido N-hexadecil-N-decanoil-para-amino-benoilsulfónico etil-diisopropilamonio (DGDASC), donde la amina hidrofóbica que contiene el grupo SO 3 -, capaz de intercambio catiónico. Después de pasar la solución modificadora, la absorción a l = 260 nm alcanzó 6,4 unidades de densidad óptica (° E) alcanzando una meseta. La capacidad de intercambio iónico calculada es de 15,65 µmol. Dado que los cationes de los elementos alcalinotérreos y el magnesio no se absorben en la región UV del espectro, se usó la detección indirecta de UV utilizando el eluyente absorbente de UV sintetizado bromuro de 1,4-dipiridinio butano (bromuro de DPB). Dado que los iones de halógeno destruyen las partes de acero de la columna, el ion bromuro del 1,4-dipiridinio butano se reemplazó por un ion acetato. Cuando la columna se lava con el eluyente, el contraión del modificador, etildiisopropilamonio, se reemplaza por el ion absorbente de UV 1,4-dipiridiniobutano. La separación de cationes se llevó a cabo a la longitud de onda óptima l = 260 nm en una escala de 0,4 A en el modo de "plegamiento de escala"; la polaridad de la grabadora estaba invertida. La separación de todos los cationes estudiados se logró con la introducción de un aditivo complejante: ácido oxálico. Los límites de detección de Mg 2+ , Ca 2+ , Sr 2+ , Ba 2+ son 8 μg/l; 16 µg/l; 34 µg/l; 72 µg/l, respectivamente. En las condiciones seleccionadas se analizó agua corriente cuyo contenido de Ca 2+ es de 10,6 +1,9 mg-ion/l, Mg 2+ -2,5 + mg-ion/l. El error de reproducibilidad no supera el -2,2 % para Ca 2+ y el 1,4 % para Mg 2+.
Análisis de complejos de cadmio en el medio ambiente
Para estudiar los mecanismos de migración de los metales pesados en la biosfera, se necesitan datos sobre las formas químicas de existencia de los metales en la naturaleza. Las dificultades en el análisis de compuestos de uno de los metales más tóxicos, el cadmio, están asociadas al hecho de que forma complejos inestables, y al intentar aislarlos se distorsionan los equilibrios naturales. En este trabajo se estudiaron los compuestos de cadmio en suelos y plantas mediante una técnica basada en la separación cromatográfica de extractos seguida de la identificación de los componentes por análisis químico. Este enfoque hizo posible no solo identificar las formas químicas del cadmio, sino también rastrear sus transformaciones en objetos ambientales.
Los grupos OH de carbohidratos y polifenoles (incluidos los flavonoides), C=O, fosfatos, NH 2 , NO 2 , los grupos SH están coordinados con el cadmio en los objetos de la biosfera. Para los fines de este estudio, se compiló un conjunto de ligandos modelo que representan estas clases de compuestos. La interacción de los ligandos modelo con las sales de cadmio solubles en agua se estudió mediante espectroscopia UV y HPLC.
Para aislar los compuestos de cadmio, se utilizó la extracción con solventes especialmente seleccionados (que no forman complejos con Cd). De esta forma, el cadmio se puede separar de todos los metales pesados, excepto de su análogo químico cercano, el zinc. Los picos que contienen cadmio y zinc en los cromatogramas de los extractos obtenidos se detectaron mediante la unión de metales en forma de sus ditizonatos. Para separar del zinc se utilizó la diferencia en la estabilidad de los complejos de Cd y Zn a pH 6-8. Los compuestos de Cd aislados se identificaron por HPLC con cambios de pH durante la elución. Se realizó el análisis de compuestos de cadmio con los componentes de suelos y tejidos vegetales, y se identificaron las sustancias producidas por las plantas en respuesta a un aumento en la ingesta de cadmio del suelo. Se ha demostrado que los flavonoides, en particular la tricina, son agentes protectores en los cereales, los alcoxi derivados de la cisteína en las leguminosas, y tanto los polifenoles como los tioles en las crucíferas.
CAPÍTULO 4. EQUIPOS DE HPLC
SERIE ACCELA
El nuevo cromatógrafo de líquidos de rendimiento ultraalto ACCELA es capaz de operar en una amplia gama de velocidades de flujo y presiones, brindando separaciones HPLC típicas en columnas convencionales y separaciones ultrarrápidas y eficientes en columnas con un tamaño de partícula adsorbente de menos de 2 µm en Presiones ultra altas (superiores a 1000 atm.).
El sistema incluye una bomba inerte de gradiente cuaternario capaz de generar presiones superiores a 1000 atm y con un volumen de retardo de solo 65 µl, lo que proporciona separaciones cromatográficas de alta velocidad. Automuestreador ACCELA capaz de operar en un ciclo de inyección de muestra de 30 segundos y proporciona la más alta reproducibilidad de inyección. Detector de red de diodos Accela PDA con un volumen de celda de flujo minimizado (2 µl) está optimizado para cromatografía de alta velocidad, utiliza tecnología LightPipe patentada y mantiene la forma de pico simétrica que viene con un sistema de cromatografía y columnas impecables.
El sistema se empareja perfectamente con los espectrómetros de masas para crear los mejores y más potentes sistemas LC/MS disponibles en el mundo.
Columnas UHP de 1,9 µm disponibles de Thermo Electron para todas las aplicaciones
SERIE TSP
El principio modular de construcción de los instrumentos de HPLC permite al cliente completar de forma flexible el equipo para resolver cualquier problema analítico y, cuando cambia, puede modificarse de forma rápida y económica. La amplia gama de módulos incluye bombas de gradiente isocrático a cuaternario, de microcolumna a semipreparativo, todos los detectores disponibles, sistemas de inyección de muestras, desde inyectores manuales hasta inyectores automáticos con la capacidad de manejar cualquier muestra, software potente para procesar resultados de medición y administrar todos los módulos del sistema. Todos los módulos están certificados según CSA, TUF/GS, FCC(EMI), VDE (EMI), ISO-9000, son compactos, tienen un diseño moderno, son fáciles de operar, están equipados con una pantalla integrada y auto- sistema de diagnóstico, le permite crear y almacenar métodos de tareas en parámetros de memoria. Cumplen con los criterios de "Práctica de laboratorio ejemplar" (GLP) y están incluidos en el Registro de instrumentos de medición de la Federación Rusa. Los protocolos de medición se emiten de acuerdo con las Farmacopeas de Inglaterra, EE. UU., Alemania y Francia.
Los sistemas modulares TSP se caracterizan por la máxima fiabilidad y estabilidad de funcionamiento.
La combinación de módulos proporciona al analista todas las ventajas de un sistema integrado por un lado y la flexibilidad de un sistema modular por el otro. Cualquiera que sea el campo de aplicación de la cromatografía líquida de alta resolución (HPLC) (farmacología, biotecnología, análisis ambiental, análisis clínico, análisis de alimentos y bebidas, análisis petroquímico y químico), este instrumento se utiliza, siempre está configurado de manera óptima para cumplir con los requisitos más exigentes. .
Tanto los sistemas de rutina de investigación como los de alto rendimiento proporcionan:
Desgasificación de solventes altamente eficiente
Capacidad para trabajar con cantidades de muestra pequeñas y ultrapequeñas
Máxima sensibilidad, tanto con detector UV/VIS como con matriz de diodos (con la famosa tecnología LightPipe con opción de longitud de camino óptico de 1 o 5 cm)
Trabajando con diferentes columnas
Precisión cuantitativa más alta
Posibilidad de trabajo automático con diferentes volúmenes de muestra
Error de tiempo de retención RMS inferior al 0,3 %
El área mínima de trabajo ocupada por el sistema.
Máxima fiabilidad y estabilidad de parámetros.
Bomba Surveyor LC- Una bomba HPLC con la mejor reproducibilidad del tiempo de retención de cualquier bomba de gradiente de 4 componentes disponible en el mundo. Un desgasificador de vacío de canal cuádruple integrado y un amortiguador de pulsaciones proporcionan una excelente estabilidad de línea de base para lograr la máxima precisión y sensibilidad de cuantificación.
El muestreador automático proporciona el máximo rendimiento y flexibilidad de análisis. Una amplia gama de bandejas de muestras, desde viales estándar hasta microplacas de 96 y 384 pocillos, cubre las necesidades de prácticamente todas las aplicaciones. La nueva tecnología proporciona prácticamente ninguna pérdida de inyección de muestra, se inyectan prácticamente 5 µl de muestra con un muestreador automático de un volumen total de muestra de 5 µl.
TOPÓGRAFO
Detector UV/Visual y PDA (Detector de Matriz de Diodos)
Topógrafo UV/Vis- El detector de luz visible y ultravioleta de longitud de onda variable es una combinación de economía y confiabilidad con la más alta sensibilidad de la tecnología LightPipe. Una amplia selección de celdas de flujo hace que este detector sea versátil para todas las aplicaciones, desde aquellas que usan cromatografía capilar o de microcolumna hasta semipreparativas y preparativas.
Topógrafo PDA El detector es el más sensible entre todos los detectores de matriz de diodos HPLC. La óptica de fuente de lámpara dual cubre perfectamente todo el rango de longitud de onda de 190 a 800 nm. El formador de haz de fibra óptica proporciona una excelente resolución óptica sin sacrificar la sensibilidad.
Agrimensor RI detector refractométrico con cubeta termostatizada de mínimo volumen con control electrónico completo desde un ordenador.
Topógrafo Florida detector de barrido fluorométrico con la más alta sensibilidad y capacidad de detección para fluorescencia, quimioluminiscencia y fosforescencia.
Una amplia gama de muestreadores automáticos le permite trabajar tanto con viales convencionales como con placas de 96 posiciones, ampliamente utilizadas en bioquímica y práctica clínica. El manejo se ve facilitado por el uso de placas de preparación de muestras SPE similares.
400 Accionamiento eléctrico, bucle Valco (20 µl - estándar) con posibilidad de llenado parcial.
Carrusel 96 muestras.
Accionamiento eléctrico, termostato de columna, bucle Valco (100 µl - estándar) con posibilidad de llenado parcial Modo AutoMix para preparación de muestras. Carrusel de muestras: 84 x 2 ml (muestras) + 3 x 10 ml (reactivos). Termostato de columna incorporado. 420
Automuestreador de bucle para trabajos de investigación con la capacidad de trabajar en los modos de llenado total, parcial e introducción de muestras de microlitros. Una amplia gama de carruseles (estándar - 96 muestras).
Automuestreador de tabletas para placas de 96 y 384 posiciones. Inyección de la muestra en el circuito de presión, posibilidad de introducir muestras de menos de 1 µl. Posibilidad de instalar un alimentador de tabletas. HPLC
Principales fabricantes de equipos HPLC
· Aguas - cromatografía de alta resolución, espectrometría de masas, columnas, extracción en fase sólida;
varian, inc. - cromatógrafos y columnas, accesorios para extracción en fase sólida;
· Agilent Technologies - cromatógrafos y columnas;
· Hypersil - columnas y adsorbentes.
· Merck KGaA - Placas TLC y accesorios para TLC, columnas, adsorbentes, fases móviles para HPLC, accesorios para extracción en fase sólida
· Dionex - equipos y columnas para HPLC, especialmente para cromatografía iónica.
Literatura
1.Pilipenko A.T., Pyatnitsky I.V. Química analítica. En dos libros: kn..1 - M .: Química, 1990, -480s.
1. Pilipenko A.T., Pyatnitsky I.V. Química analítica. En dos libros: kn..2 - M .: Química, 1990, -480s.
2. Vasiliev V.P. Química analítica. A las 14 h Parte 2. Métodos físicos y químicos de análisis: Proc. para Khimko - tecnología. especialista. universidades - M.: Superior. escuela, 1989. - 384p.
3. Materiales hidroquímicos. Tomo 100. Métodos y medios técnicos de vigilancia operativa de la calidad de las aguas superficiales. L.: Gidrometeo-izdat, 1991. - 200p.
4. Lurie Yu.Yu. Química analítica de aguas residuales industriales / Yu.Yu. Lurie; M.: Química Yu, 1984. - 448s.
5. Ewing G. Métodos instrumentales de análisis químico / Per. De inglés. M.: Mir, 1989. - 348 p.
6. Gorelik DO, Konopelko L.A., Pankov E.D. Monitoreo ambiental. En 2 volúmenes San Petersburgo: Navidad. 2000. - 260 págs.
7. Aivazov B.V. Introducción a la cromatografía. M.: Superior. escuela, 1983. - 450 p.
8. Goldberg K.A., Vigdergauz M.S. Introducción a la cromatografía de gases. M.: Química, 1990. - 329 p.
9. Stolyarov B.V. y otros // Prácticas de cromatografía de gases y líquidos. San Petersburgo: Universidad Estatal de San Petersburgo, 1998. - S. 81.
11. Gorshkov A.G., Marinaite I.I. HPLC: un método para monitorear PAH en objetos ambientales
12. Torosyan G. O., Martirosyan V. A., Aleksanyan A. R., Zakaryan M. O. Eliminación de anilina de soluciones acuosas utilizando productos de desecho de reducción aluminotérmica de incrustaciones de cobre laminado
13. Los Ángeles Turkina, G. N. Koroleva Desarrollo de un método para la determinación de elementos alcalinotérreos y magnesio por cromatografía líquida iónica de alta resolución
14. Dultseva G.G., Dubtsova Yu.Yu., Skubnevskaya G.I. Análisis de complejos de cadmio en el medio ambiente
Apéndice
DETERMINACIÓN DE CLOMAZONA EN AGUA POR MÉTODOS CROMATOGRAFICOS
INSTRUCCIONES METODOLÓGICAS MUK 4.1.1415-03
1. Elaborado por: Centro Científico Federal de Higiene. F. F.
Erismán; Academia Agrícola de Moscú. K. A.
Timiryazev; con la participación del Departamento Estatal de Vigilancia Sanitaria y Epidemiológica del Ministerio de Salud de Rusia. Los desarrolladores de la metodología se enumeran al final.
3. Aprobado por el Médico Jefe de Sanidad del Estado
Federación Rusa, Primer Viceministro de Salud de la Federación Rusa, acad. RAMS G.G. Onishchenko 24 de junio de 2003
5. Presentado por primera vez.
1. Introducción
Fabricante: FMS (EE.UU.).
Nombre comercial: COMANDO.
Principio activo: clomazona.
2-(2-clorobencil)-4,4-dimetil-3-isoxalidin-3-ona (IUPAC)
Líquido viscoso de color marrón claro.
Punto de fusión: 25 -C.
Punto de ebullición: 275 -C.
Presión de vapor a 25 -C: 19,2 MPa.
Coeficiente de reparto n-octanol/agua: K logP = 2,5.
Altamente soluble en acetona, hexano, etanol, metanol,
cloroformo, diclorometano y acetonitrilo; solubilidad en agua -
1,10 g/pie DM Estable a temperatura ambiente durante al menos 2 años, a 50 -C - al menos 3 meses.
Breve perfil toxicológico: Oral aguda
toxicidad (LD) para ratas - 1369 - 2077 mg/kg; dérmica aguda
toxicidad (LD) para ratas - más de 2000 mg/kg; agudo
toxicidad por inhalación (LC) para ratas - 4,8 mg / cu. dm (4 horas).
Normas higiénicas. MPC en agua - 0,02 mg / cu. DM
Alcance de la droga. Clomazone es un herbicida selectivo utilizado para el control de cereales y malas hierbas dicotiledóneas en cultivos de soja y arroz durante la aplicación previa a la emergencia o la siembra.
2. Método para la determinación de clomazona en agua.
métodos cromatográficos
2.1. Puntos clave
2.1.1. El principio de la técnica.
La técnica se basa en la extracción de clomazone de la muestra analizada con hexano, concentración del extracto y posterior determinación cuantitativa por métodos alternativos:
cromatografía líquida de alta resolución (HPLC) con
detector ultravioleta, cromatografía gas-líquido (GLC) con un detector de velocidad de recombinación constante o cromatografía en capa fina (TLC). La determinación cuantitativa se lleva a cabo por el método de calibración absoluta.
2.1.2. Selectividad del método
En las condiciones propuestas, el método es específico en presencia de contaminantes ambientales globales: derivados clorados de cicloparafinas (isómeros de HCH), compuestos de difenilo (DDT y sus derivados), sus metabolitos - bencenos policlorados y fenoles, así como en presencia de tricloroacetato de sodio, que se puede utilizar en cultivos como herbicida.
2.1.3. Característica metrológica del método (P = 0,95)
Reactivos, soluciones y materiales.
Clomazone con el contenido de d. 99,8%
(FMS, EE. UU.)
Nitrógeno, y GOST 9293-79
Agua amoníaco, 25%, h GOST 1277-81
Acetona, h GOST 2603-79
n-Hexano, h GOST 2603-79
Peróxido de hidrógeno, solución acuosa al 30% GOST 10929-77
Alcohol isopropílico, químicamente puro TU 6-09-402-75
Ácido sulfúrico, químicamente puro GOST 4203-77
Ácido clorhídrico (clorhídrico), químicamente puro GOST 3118-77
Alcohol metílico, GOST químicamente puro
Hidróxido de sodio, químicamente puro, solución acuosa al 25% GOST 4323-77
Sulfato de sodio anhidro, químicamente puro GOST 1277-81
Nitrato de plata, químicamente puro GOST 1277-81
2-fenoximetanol, h TU 6-09-3688-76
Chromaton N-AW-DMCS (0,16 - 0,20 mm)
con 5% SE-30, Hemapol, República Checa
Chromaton N-AW-DMCS (0,16 - 0,20 mm) con 1,5
OV-17 + 1,95% QF-1, Hemapol, República Checa
Placas para HPTLC (URSS)
Registros "Kieselgel 60 F-254" (Alemania)
Registros "Silufol" República Checa
Filtros de papel "cinta blanca", sin cenizas y prelavado con hexano TU 6-09-2678-77
2.3. Cubiertos, equipo, utensilios
Cromatógrafo de líquidos Milichrome
con detector ultravioleta
Columna cromatográfica de acero,
longitud 64 mm, diámetro interior 2 mm,
relleno con Silasorb 600, tamaño de grano 5 µm
Cromatógrafo de gases serie "Color" o
similar, equipado con un detector constante
tasa de recombinación (RPR) con un límite
detección por lindano 4 x 10 g/cu. cm
Columna de vidrio cromatográfico, longitud
1 o 2 m, diámetro interior 2 - 3 mm
Microjeringa tipo MSH-10, capacidad 10 µl TU 5E2-833-024
Aparato de agitación tipo AVU-6s TU 64-1-2851-78
Baño de agua TU 64-1-2850-76
Balanza analítica tipo VLA-200 GOST 34104-80E
Cámara cromatográfica GOST 10565-74
Bomba de chorro de agua GOST 10696-75
Irradiador de mercurio-cuarzo tipo OKN-11 TU 64-1-1618-77
Pistolas de vidrio GOST 10391-74
Evaporador rotativo al vacío IR-1M
o similar TU 25-11-917-76
Unidad compresora TU 64-1-2985-78
Armario de secado TU 64-1-1411-76E
Embudos divisorios GOST 3613-75
Matraces volumétricos, con una capacidad de 100 ml GOST 1770-74
Probetas graduadas, con una capacidad de 10, 50 ml GOST 1770-74E
Frascos en forma de pera con una sección delgada,
con una capacidad de 100 ml GOST 10394-72
Matraces cónicos, con una capacidad de 100 ml GOST 22524-77
Tubos de ensayo de centrífuga, medidos GOST 25336-82E
Pipetas, con capacidad de 0,1, 1, 2, 5 y 10 ml GOST 20292-74
Embudos químicos, cónicos, diámetro
34 - 40 mm GOST 25336-82E
2.4. Selección de muestras
El muestreo, el almacenamiento y la preparación de las muestras se llevan a cabo de acuerdo con
"Reglas unificadas para el muestreo de productos agrícolas, productos alimenticios y objetos ambientales para la determinación de cantidades traza de pesticidas", aprobada por N 2051-79 del 21.08.79
Las muestras tomadas se pueden almacenar en el refrigerador hasta por 5 días. Antes del análisis, el agua (en presencia de suspensión) se filtra a través de un filtro de papel suelto.
2.5. Preparación para la definición
2.5.1. método HPLC
2.5.1.1. Preparación de fase móvil para HPLC
En un matraz aforado de 100 ml de capacidad se colocan con una pipeta 5 ml de isopopanol y 5 ml de metanol, se completa hasta el enrase con hexano, se mezcla, se filtra.
2.5.1.2. Acondicionamiento de columna
Enjuague la columna de HPLC con hexano-metanol-isopropanol (90:5:5, v/v) durante 30 min. a una velocidad de alimentación de disolvente de 100 µl/min.
2.5.2. método GLC. Preparación y acondicionamiento de columnas
El relleno terminado (5% SE-30 en Chromaton N-AW-DMCS) se vierte en una columna de vidrio, se compacta al vacío, la columna se instala en el cromatógrafo termostato sin conectar al detector y se estabiliza en una corriente de nitrógeno a una temperatura de 250 -C durante 10 - 12 del mediodía
2.5.3. método CCF
2.5.3.1. Preparación de reactivos de desarrollo.
2.5.3.1.1. Reactivo de revelado nº 1
Se disuelve 1 g de nitrato de plata en 1 ml de agua destilada, se agregan 10 ml de 2-fenoximetanol, 190 ml de acetona, 1-2 gotas de peróxido de hidrógeno, la solución se agita y se transfiere a una botella de vidrio oscuro.
2.5.3.2.2. Reactivo de revelado N 2
Se disuelven 0.5 g de nitrato de plata en 5 ml de agua destilada en un matraz aforado de 100 ml, se agregan 10 ml de amoníaco acuoso al 25%, se ajusta la solución a 100 ml con acetona, se mezcla y se transfiere a un frasco de vidrio oscuro.
2.5.3.2. Preparación de la fase móvil para TLC
En un matraz aforado de 100 ml de capacidad agregar 20 ml de acetona y agregar hexano hasta el enrase, mezclar. La mezcla se vierte en la cámara cromatográfica con una capa de no más de 6 - 8 mm en 30 minutos. Antes de iniciar la cromatografía.
2.5.4. Preparación de soluciones estándar
Se prepara una solución estándar madre de clomazona de 100 µg/mL disolviendo 0,010 g de una preparación que contiene 99,8 % de AI en hexano en un matraz volumétrico de 100 mL. La solución se almacena en el refrigerador durante un mes.
Soluciones estándar de trabajo con una concentración de 0,4; 1,0; 2.0; 4.0; 10,0; Se preparan 20 y 40,0 µg/ml a partir de la solución estándar madre de clomazona mediante diluciones seriadas apropiadas con hexano.
Las soluciones de trabajo se almacenan en el refrigerador por no más de un mes.
2.5.5. Construcción de un gráfico de calibración
2.5.5.1. Curva de calibración A (medición según el párrafo 2.7.1, HPLC)
Para construir un gráfico de calibración, se inyectan 5 µl de una solución estándar de trabajo de clomazone con una concentración de 4,0 en el inyector del cromatógrafo; 10,0; 20,0 y 40 µg/ml.
2.5.5.2. Curva de calibración B (medición según el párrafo 2.7.2, GLC)
Para construir un gráfico de calibración, se inyectan 5 µl de una solución estándar de trabajo de clomazone con una concentración de 0,4 en el evaporador del cromatógrafo; 1,0; 2.0; 4.0 y 10.0.
Realice al menos 5 mediciones paralelas. Encuentre la altura promedio del pico cromatográfico para cada concentración. Construya un gráfico de calibración (A o B) de la dependencia de la altura del pico cromatográfico en mm de la concentración de clomazone en solución en µg/ml.
2.6. Definición Descripción
Se colocan 100 ml de la muestra de agua analizada en un embudo de decantación de 250 ml de capacidad, se añaden 10 ml de una solución acuosa al 25% de hidróxido de sodio, se mezcla y se añaden 20 ml de n-hexano. El embudo se agita durante 3 minutos, después de la separación de fases, la capa de hexano se vierte en un matraz en forma de pera con una capacidad de 100 ml, pasándolo a través de una capa de sulfato de sodio anhidro colocado en un embudo cónico sobre un papel de filtro plegado. La extracción del fármaco de la muestra acuosa se repite dos veces más utilizando 20 ml de n-hexano. El extracto de hexano combinado se evapora en un evaporador de vacío rotatorio a una temperatura de 40 -C casi hasta la sequedad, el residuo se elimina con una corriente de aire o nitrógeno de pureza especial. El residuo seco se disuelve en 0,1 (HPLC, TLC) o 0,25 ml (GLC) de n-hexano y se analiza por uno de los métodos cromatográficos.
2.7. Condiciones de cromatografía
Cromatógrafo de líquidos con detector ultravioleta Milichrom (Rusia).
Columna de acero de 64 mm de largo, diámetro interior de 2 mm,
lleno de Silasorb 600, granulometría 5 micras.
Temperatura de la columna: ambiente.
Fase móvil: hexano-isopropanol-metanol (90:5:5, v/v).
Caudal de eluyente: 100 µl/min.
Longitud de onda de funcionamiento: 240 nm.
Sensibilidad: 0,4 unidades absorción en la escala.
Volumen de inyección: 5 µl.
Tiempo de salida de Clomazone: unos 6 min.
Rango de detección lineal: 20 - 200 ng.
Las muestras que producen picos superiores a la solución estándar de 40 µg/ml se diluyen con fase móvil de HPLC.
Cromatógrafo de gases "Tsvet-570" con detector de tasa de recombinación de iones constante.
Columna de vidrio de 1 m de largo, 3 mm de diámetro interior, rellena con Chromaton N-AW-DMCS con 5% SE-30 (0,16 - 0,20 mm).
La escala de trabajo del electrómetro es 64 x 10 10 Ohm.
Grabadora de velocidad de cinta 200 mm/h.
Temperatura del termostato de columna - 190 -C
detector - 300 -C
evaporador - 220 -C
La velocidad del gas portador (nitrógeno) - 60 ml / min.
El volumen de la muestra inyectada es de 5 µl.
El tiempo de salida de la clomazona es de 2,5 minutos.
Rango de detección lineal: 2 - 50 ng.
Las muestras que producen picos mayores que la solución estándar de 10 µg/mL se diluyen con hexano.
Para mejorar la precisión de la identificación de la clomazona en presencia de gamma-HCCH que tiene un tiempo de retención cercano en la muestra, la clomazona se elimina de la muestra mediante tratamiento con ácido sulfúrico concentrado. El nuevo análisis de la muestra le permite establecer la contribución de la clomazona a la señal cromatográfica primaria.
Solución de hexano en un matraz, obtenido de acuerdo con el párrafo 2.6 cuantitativamente
(o una alícuota del mismo) se aplica a las placas cromatográficas "Silufol", "Kieselgel 60F-254" o "Plates for HPTLC". Cerca, las soluciones estándar se aplican en un volumen correspondiente al contenido de clomazone 1, 2, 5 y 10 μg. La placa se coloca en una cámara cromatográfica que contiene una mezcla de n-hexano-acetona (4:1, v/v). Después del revelado del cromatograma, la placa se retira de la cámara, se coloca bajo corriente de aire hasta que se evaporen los solventes, luego se trata con uno de los reactivos de revelado y se coloca bajo una lámpara ultravioleta durante 5 minutos. La zona de localización del fármaco en las placas "Silufol", "Placas para HPTLC" y "Kieselgel 60F-254" aparece como manchas de color marrón grisáceo con un valor Rf de 0,35, 0,85 y 0,43, respectivamente. Para determinar la clomazona por TLC, puede usar placas "Alugram" y "Polygram" (fabricadas por Alemania). El valor Rf de la clomazona en estas placas es 0,37 y 0,38, respectivamente.
3. Requisitos de seguridad
Es necesario seguir las reglas de seguridad generalmente aceptadas cuando se trabaja con solventes orgánicos, sustancias tóxicas, calentadores eléctricos.
4. Control de errores de medición
El control operativo del error y la reproducibilidad de las mediciones se lleva a cabo de acuerdo con las recomendaciones de MI 2335-95. GSI "Control de calidad interno de los resultados de análisis químicos cuantitativos".
5. Desarrolladores
Yudina T.V., Fedorova N.E. (FNTSG llamado así por FF Erisman).
Davidyuk E. I. (UkrNIIGINTOX, Kiev); Kisenko M.A., Demchenko V.F. (Instituto de Medicina del Trabajo de la Academia de Ciencias y la Academia de Ciencias Médicas de Ucrania, Kiev).
Cromatografía de adsorción de líquidos en columna
La separación de una mezcla de sustancias en una columna de adsorción se produce como resultado de su diferencia de capacidad de absorción en un adsorbente dado (de acuerdo con la ley de sustitución de adsorción establecida por M. S. Tsvet).
Los adsorbentes son cuerpos porosos con una superficie interna muy desarrollada que retienen líquidos a través de fenómenos intermoleculares y superficiales. Estos pueden ser compuestos inorgánicos y orgánicos polares y no polares. Los adsorbentes polares incluyen gel de sílice (dióxido de silicio gelatinoso seco), óxido de aluminio, carbonato de calcio, celulosa, almidón, etc. Adsorbentes no polares: carbón activado, polvo de caucho y muchos otros obtenidos sintéticamente.
Los adsorbentes están sujetos a los siguientes requisitos: S no deben entrar en reacciones químicas con la fase móvil y las sustancias a separar; S debe tener resistencia mecánica; Los granos S del adsorbente deben tener el mismo grado de dispersión.
Al elegir las condiciones para el proceso cromatográfico, se tienen en cuenta las propiedades del adsorbente y las sustancias adsorbidas.
En la versión clásica de la cromatografía en columna líquida (LCC), se pasa un eluyente (PF) a través de una columna cromatográfica, que es un tubo de vidrio de 0,5 a 5 cm de diámetro y 20 a 100 cm de largo, lleno de un adsorbente (NP). El eluyente se mueve bajo la influencia de la gravedad. La velocidad de su movimiento se puede ajustar mediante la grúa en la parte inferior de la columna. La mezcla a analizar se coloca en la parte superior de la columna. A medida que la muestra se mueve a través de la columna, los componentes se separan. A determinados intervalos se toman fracciones del eluyente liberado de la columna, las cuales son analizadas por cualquier método que permita medir las concentraciones de analitos.
La cromatografía de adsorción en columna se usa actualmente principalmente no como un método de análisis independiente, sino como un método de separación preliminar (a veces final) de mezclas complejas en otras más simples, es decir, prepararse para el análisis por otros métodos (incluido el cromatográfico). Por ejemplo, se separa una mezcla de tocoferoles en una columna de alúmina, se pasa el eluyente y se recoge la fracción de α-tocoferoles para su posterior determinación fotométrica.
La separación cromatográfica de la mezcla en la columna debido al lento avance del PF lleva mucho tiempo. Para acelerar el proceso, la cromatografía se realiza bajo presión. Este método se llama Cromatografía Líquida de Alta Resolución (HPLC)
La modernización del equipo utilizado en la cromatografía en columna líquida clásica lo ha convertido en uno de los métodos de análisis más prometedores y modernos. La cromatografía líquida de alta resolución es un método conveniente para la separación, el aislamiento preparativo y el análisis cualitativo y cuantitativo de compuestos termolábiles no volátiles de bajo y alto peso molecular.
Según el tipo de sorbente utilizado, este método utiliza 2 opciones de cromatografía: en un sorbente polar que utiliza un eluyente no polar (opción de fase directa) y en un sorbente no polar que utiliza un eluyente polar, el llamado de fase inversa alta. -cromatografía líquida de alto rendimiento (RP HPLC).
Cuando el eluyente pasa al eluyente, el equilibrio en condiciones de RPHLC se establece muchas veces más rápido que en las condiciones de adsorbentes polares y PF no acuosos. Como resultado de esto, además de la conveniencia de trabajar con agua y eluyentes hidroalcohólicos, RPHLC ahora ha ganado gran popularidad. La mayoría de los análisis de HPLC se llevan a cabo utilizando este método.
Instrumentación para HPLC
Un conjunto de equipos modernos para HPLC, por regla general, consta de dos bombas 3,4 (Fig. 7.1.1.1), controladas por un microprocesador 5 y que suministran eluyente de acuerdo con un programa específico. Las bombas crean una presión de hasta 40 MPa. La muestra se inyecta a través de un dispositivo especial (inyector) 7 directamente en el flujo de eluyente. Después de pasar por la columna cromatográfica 8, las sustancias son detectadas por un detector de flujo altamente sensible 9, cuya señal es registrada y procesada por el microordenador 11. Si es necesario, las fracciones se seleccionan automáticamente en el momento de la salida máxima.
Las columnas para HPLC están hechas de acero inoxidable con un diámetro interior de 2 a 6 mm y una longitud de 10 a 25 cm Las columnas se llenan con un adsorbente (NF). Como NF se utilizan gel de sílice, alúmina o adsorbentes modificados. El gel de sílice generalmente se modifica mediante la introducción química de varios grupos funcionales en su superficie.
detectores El registro de la salida de la columna de un componente separado se realiza mediante un detector. Para el registro, puede utilizar el cambio en cualquier señal analítica proveniente de la fase móvil y relacionada con la naturaleza y cantidad del componente de la mezcla. La cromatografía líquida utiliza señales analíticas como la absorción de luz o la emisión de luz de la solución que sale (detectores fotométricos y fluorimétricos), índice de refracción (detectores refractométricos), conductividad eléctrica y potencial (detectores electroquímicos), etc.
La señal detectada continuamente es registrada por el registrador. Un cromatograma es una secuencia de señales de detector registradas en una grabadora, que se generan cuando los componentes individuales de una mezcla salen de la columna. En el caso de separación de la mezcla, los picos separados son visibles en el cromatograma externo. La posición del pico en el cromatograma se utiliza con fines de identificación de la sustancia, la altura o el área del pico se utiliza con fines de determinación cuantitativa.
Analisis cualitativo
Las características más importantes del cromatograma - el tiempo de retención tR y el volumen de retención asociado al mismo - reflejan la naturaleza de las sustancias, su capacidad de sorción sobre el material de la fase estacionaria y, por tanto, en condiciones cromatográficas constantes, son un medios para identificar la sustancia. Para una columna determinada con un caudal y una temperatura determinados, el tiempo de retención de cada compuesto es constante (Fig. 7.1.1.2), donde tR(A) es el tiempo de retención del componente A de la mezcla analizada desde que se inyecta en la columna hasta que aparezca el pico máximo a la salida de la columna, 1K( ss) - tiempo de retención del patrón interno (sustancia inicialmente ausente en la mezcla analizada), h - altura del pico (mm), ab - anchura del pico a la mitad de su altura , mm.
Para identificar una sustancia por cromatograma, se suelen utilizar muestras estándar o sustancias puras. Compare el tiempo de retención del componente IR* desconocido con el tiempo de retención IRCT de las sustancias conocidas. Pero una identificación más confiable al medir el tiempo de retención relativo
En este caso, primero se introduce una sustancia conocida (estándar interno) en la columna y se mide su tiempo de retención tR(Bc), luego la mezcla de prueba se separa cromatográficamente (cromatografía), a la que se le agrega preliminarmente el estándar interno. El tiempo de retención relativo está determinado por la fórmula (7.1.1.1).
Análisis cuantitativo
Este análisis se basa en la dependencia de la altura del pico ho su área S de la cantidad de sustancia. Para picos estrechos, es preferible medir h, para picos anchos y borrosos - S. El área del pico se mide de diferentes maneras: multiplicando la altura del pico (h) por su ancho (ai / 2), medido a la mitad de su altura (Fig. 7.2.3); planificación; utilizando un integrador. Los cromatógrafos modernos están equipados con integradores eléctricos o electrónicos.
Se utilizan principalmente tres métodos para determinar el contenido de sustancias en una muestra: el método de calibración absoluta, el método de normalización interna y el método de patrón interno.
El método de calibración absoluta se basa en una determinación preliminar de la relación entre la cantidad de sustancia introducida y el área o altura del pico en el cromatograma. Se introduce una cantidad conocida de la mezcla de calibración en el cromatograma y se determinan las áreas o alturas de los picos resultantes. Construya un gráfico del área o la altura del pico a partir de la cantidad de sustancia inyectada. Se analiza la muestra de ensayo, se mide el área o altura del pico del componente a determinar y se calcula su cantidad en base a la curva de calibración.
Este método proporciona información solo sobre el contenido relativo del componente en la mezcla, pero no permite determinar su valor absoluto.
El método del estándar interno se basa en la comparación de un parámetro de pico seleccionado de un analito con el mismo parámetro de una sustancia estándar introducida en la muestra en una cantidad conocida. Se introduce una cantidad conocida de dicha sustancia estándar en la muestra de prueba, cuyo pico está suficientemente bien separado de los picos de los componentes de la mezcla de prueba.
Los dos últimos métodos requieren la introducción de factores de corrección que caractericen la sensibilidad de los detectores utilizados a las sustancias analizadas. Para diferentes tipos de detectores y diferentes sustancias, el coeficiente de sensibilidad se determina experimentalmente.
La cromatografía de adsorción de líquidos también utiliza el análisis de fracciones de soluciones recolectadas en el momento en que la sustancia sale de la columna. El análisis puede llevarse a cabo por varios métodos fisicoquímicos.
La cromatografía de adsorción de líquidos se utiliza principalmente para la separación de sustancias orgánicas. Este método tiene mucho éxito en el estudio de la composición del aceite, los hidrocarburos, la separación efectiva de isómeros trans y cis, alcaloides, etc. La HPLC se puede utilizar para determinar tintes, ácidos orgánicos, aminoácidos, azúcares, impurezas de pesticidas y herbicidas, sustancias medicinales. y otros contaminantes en los productos alimenticios.
(principalmente intermolecular) en la interfase. Como método de análisis, la HPLC forma parte de un grupo de métodos que, debido a la complejidad de los objetos en estudio, incluye la separación preliminar de la mezcla compleja inicial en otras relativamente simples. Las mezclas simples obtenidas se analizan luego por métodos fisicoquímicos convencionales o por métodos especiales desarrollados para la cromatografía.
El método HPLC se usa ampliamente en campos como la química, la petroquímica, la biología, la biotecnología, la medicina, el procesamiento de alimentos, la protección del medio ambiente, la producción de medicamentos y muchos otros.
Según el mecanismo de separación de las sustancias analizadas o separadas, la HPLC se divide en adsorción, distribución, intercambio iónico, exclusión, intercambio de ligandos y otros.
Debe tenerse en cuenta que en el trabajo práctico, la separación a menudo se produce no por uno, sino por varios mecanismos simultáneamente. Por lo tanto, la separación por exclusión puede complicarse por los efectos de adsorción, adsorción - distribución y viceversa. Al mismo tiempo, cuanto mayor sea la diferencia de sustancias en la muestra en términos de grado de ionización, basicidad o acidez, en términos de peso molecular, polarizabilidad y otros parámetros, más probable es que aparezca un mecanismo de separación diferente. para tales sustancias.
HPLC en fase normal
La fase estacionaria es más polar que la fase móvil, por lo que el disolvente no polar predomina en la composición del eluyente:
- Hexano:isopropanol = 95:5 (para sustancias poco polares)
- Cloroformo:metanol = 95:5 (para sustancias polares medias)
- Cloroformo:metanol = 80:20 (para sustancias altamente polares)
HPLC de fase inversa
La fase estacionaria es menos polar que la fase móvil, por lo que casi siempre hay agua en el eluyente. En este caso, siempre es posible asegurar la disolución completa del BAS en la fase móvil, casi siempre es posible usar la detección UV, casi todas las fases móviles son miscibles entre sí, se puede usar elución en gradiente, la columna se puede volver a cambiar rápidamente -equilibrada, la columna se puede regenerar.
Los eluyentes comunes para HPLC de fase inversa son:
- Acetonitrilo: agua
- metanol: agua
- Isopropanol: agua
Matrices para HPLC
Las matrices utilizadas en HPLC son compuestos inorgánicos como sílice (gel de sílice) o alúmina, o polímeros orgánicos como poliestireno (reticulado con divinilbenceno) o polimetacrilato. El gel de sílice es, por supuesto, ahora generalmente aceptado.
Las principales características de la matriz:
- Tamaño de partícula (µm);
- Tamaño de poro interno (Å, nm).
Obtención de gel de sílice para HPLC:
- Formación de microesferas de ácido polisilícico;
- Secado de partículas de gel de sílice;
- Separación de aire.
Partículas absorbentes:
- Regular (esférica): mayor resistencia a la presión, mayor costo;
- No esférica: menor resistencia a la presión.
El tamaño de poro en HPLC es uno de los parámetros más importantes. Cuanto menor sea el tamaño de los poros, peor será su permeabilidad para las moléculas de las sustancias eluidas. Y en consecuencia, peor será la capacidad de sorción de los adsorbentes. Cuanto más grandes son los poros, menor es, en primer lugar, la estabilidad mecánica de las partículas adsorbentes y, en segundo lugar, cuanto menor es la superficie de sorción, por lo tanto, peor es la eficiencia.
Injertos de fase estacionaria
HPLC en fase normal:
- Fase estacionaria injertada con propilnitrilo (nitrilo);
- Fase estacionaria con injerto de propilamina (amina).
HPLC de fase inversa:
- Fase estacionaria con injerto de alquilo;
- Fase estacionaria con injerto de alquilsililo.
Recubrimiento final: protección de áreas no injertadas del sorbente mediante injertos adicionales con moléculas "pequeñas". Protección terminal hidrofóbica (C1, C2): mayor selectividad, peor humectabilidad; protección terminal hidrófila (diol): menor selectividad, mayor humectabilidad.
detectores de HPLC
- ultravioleta
- matriz de diodos
- Fluorescente
- electroquímico
- refractométrico
- selectivo de masas
Enlaces
Fundación Wikimedia. 2010 .
Vea qué es "Cromatografía líquida de alto rendimiento" en otros diccionarios:
cromatografía líquida de alta resolución- - [AS Goldberg. Diccionario de energía inglés ruso. 2006] Temas energía en general EN cromatografía líquida de alta resoluciónHPLC … Manual del traductor técnico
Término cromatografía líquida de alta resolución Término inglés cromatografía líquida de alta resolución Sinónimos Abreviaturas HPLC, HPLC Términos relacionados adsorción, oligopéptido, proteómica, sorbente, fullereno, endoédrico, cromatografía… …
Cromatografía líquida, para aumentar la eficiencia de la separación, el solvente (eluyente) bajo presión (más de 3x107 Pa) se bombea a través de columnas llenas de un sorbente con partículas de pequeño diámetro (hasta 1 μm) y perfusión ... ...
Un tipo de cromatografía en la que el líquido (eluyente) sirve como fase móvil y es la fase estacionaria. absorbente, tv. un portador con un líquido o gel aplicado a su superficie. Se lleva a cabo en una columna rellena con un adsorbente (cromatografía en columna), en un plano ... ... Ciencias Naturales. diccionario enciclopédico
- [κρώμα (υroma) color] un proceso basado en la capacidad desigual de los componentes individuales de la mezcla (líquidos o gaseosos) para permanecer en la superficie del adsorbente tanto cuando son absorbidos de la corriente portadora como cuando... ... Enciclopedia geológica
- (de otro griego ... Wikipedia
Cromatografía de términos Cromatografía de términos en inglés Sinónimos Abreviaturas Términos asociados cromatografía líquida de alta resolución, clatrato, laboratorio en un chip, porosimetría, proteoma, proteómica, sorbente, enzima, fullereno, endoédrico… … Diccionario Enciclopédico de Nanotecnología
Cromatografía líquida basada en descomp. la capacidad de los iones separados de intercambiar iones con los fijos. iones absorbentes formados como resultado de la disociación de los grupos ionogénicos de este último. Los intercambiadores de cationes se utilizan para separar cationes, para ... ... Enciclopedia química
HPLC- cromatografía líquida de alta resolución ... Diccionario de abreviaturas del idioma ruso
La cromatografía líquida de alta resolución (HPLC) es uno de los métodos efectivos para separar mezclas complejas de sustancias, que se usa ampliamente tanto en química analítica como en tecnología química. La base de la separación cromatográfica es la participación... Wikipedia
Libros
- Práctica de cromatografía líquida de alta resolución, Veronica R. Mayer. Presentamos al lector la 5ª edición del libro, que se ha ampliado con métodos y equipos modernos. Se ha mejorado mucho en el libro y se ha añadido un gran número de referencias. Los lugares del texto donde...
9885 0
La HPLC es una cromatografía en columna líquida, en la que se pueden utilizar una variedad de mecanismos de sorción. Esencialmente, HPLC es una forma moderna de cromatografía en columna líquida clásica. A continuación se enumeran algunas de las características cualitativas más significativas de HPFA:
- alta velocidad del proceso, que permitió reducir la duración de la separación de varias horas y días a minutos;
- el mínimo grado de borrosidad de las zonas cromatográficas, que permite separar compuestos que difieren sólo ligeramente en las constantes de sorción;
- un alto grado de mecanización y automatización de la separación y procesamiento de la información, gracias al cual la cromatografía líquida en columna ha alcanzado un nuevo nivel de reproducibilidad y precisión.
Estudios intensivos de las últimas décadas, una gran cantidad de datos experimentales acumulados permiten hoy hablar sobre la clasificación de variantes en el marco del método de cromatografía líquida de alta resolución. Por supuesto, en este caso, la clasificación según el mecanismo de sorción dada anteriormente sigue siendo válida.
Una clasificación común se basa en la polaridad comparativa de las fases móvil y estacionaria. Se hace una distinción entre cromatografía de fase normal y reversa.
La cromatografía en fase normal (NPC) es una variante de HPLC cuando la fase móvil es menos polar que la fase estacionaria, y hay razones para creer que el factor principal que determina la retención es la interacción de los sorbatos directamente con la superficie o el volumen del adsorbente. .
La cromatografía de fase inversa (RPC) es una variante de la HPLC cuando la fase móvil es más polar que la estacionaria y la retención está determinada por el contacto directo de las moléculas de sorbato con la superficie o volumen del adsorbente; en este caso, los sorbatos ionizados no se intercambian por iones de la fase móvil adsorbidos en la superficie.
Cromatografía de intercambio iónico: una variante en la que la sorción se lleva a cabo mediante el intercambio de iones absorbidos de la fase móvil por iones de sustancias cromatográficas; la cromatografía de intercambio de ligando se puede determinar exactamente de la misma manera.
La cromatografía en sorbentes modificados dinámicamente es una variante de HPLC, en la que el sorbato no interactúa directamente con la superficie del sorbente, sino que entra en asociación con las moléculas de las capas cercanas a la superficie del eluyente.
La cromatografía de pares de iones es una variante de la cromatografía de fase inversa de compuestos ionizados, en la que se agrega un contraión hidrofóbico a la fase móvil, lo que cambia cualitativamente las características de sorción del sistema.
La cromatografía de exclusión por tamaño es un método para separar compuestos por sus pesos moleculares, basado en la diferencia en la tasa de difusión en los poros de la fase estacionaria de moléculas de varios tamaños.
Para HPFA, una característica muy importante es el tamaño de los sorbentes, generalmente de 3 a 5 µm, ahora hasta 1,8 µm. Esto permite separar mezclas complejas de sustancias de forma rápida y completa (el tiempo medio de análisis es de 3 a 30 min).
El problema de la separación se resuelve utilizando una columna cromatográfica, que es un tubo lleno de un adsorbente. Durante el análisis, un líquido (eluyente) de cierta composición se alimenta a través de una columna cromatográfica a una velocidad constante. En esta corriente se inyecta una dosis de muestra medida con precisión. Los componentes de la muestra introducidos en la columna cromatográfica, debido a su diferente afinidad con el sorbente de la columna, se desplazan a lo largo de ésta a distintas velocidades y llegan al detector secuencialmente en distintos momentos.
Así, la columna cromatográfica es responsable de la selectividad y eficiencia de separación de los componentes. Al seleccionar diferentes tipos de columnas, puede controlar el grado de separación de las sustancias analizadas. Los compuestos se identifican por su tiempo de retención. La determinación cuantitativa de cada uno de los componentes se calcula en base a la magnitud de la señal analítica medida mediante un detector conectado a la salida de la columna cromatográfica.
Sorbentes. La formación de HPLC está asociada en gran medida con la creación de nuevas generaciones de adsorbentes con buenas propiedades cinéticas y diversas propiedades termodinámicas. El material principal para los adsorbentes en HPLC es el gel de sílice. Es mecánicamente fuerte y tiene una porosidad significativa, lo que le otorga una gran capacidad de intercambio para tamaños de columna pequeños. El tamaño de partícula más común es de 5 a 10 micrones. Cuanto más se acerque a la forma esférica de las partículas, menor será la resistencia al flujo, mayor será la eficiencia, especialmente si se filtra una fracción muy estrecha (por ejemplo, 7 +1 micras).
El área de superficie específica del gel de sílice es de 10-600 m/g. El gel de sílice se puede modificar con varios grupos químicos injertados en la superficie (C-18, CN, NH2, SO3H), lo que hace posible usar adsorbentes basados en él para separar varias clases de compuestos. La principal desventaja del gel de sílice es su baja resistencia química a pH< 2 и рН >9 (el sílice se disuelve en álcalis y ácidos). Por ello, actualmente se está realizando una intensa búsqueda de sorbentes basados en polímeros que sean estables a pH de 1 a 14, por ejemplo, a base de polimetilmetacrilato, poliestireno, etc.
Sorbentes para cromatografía de intercambio iónico. Debido a las peculiaridades de separación (en medio ácido o alcalino), el material principal es sorbente a poliestireno con divinilbenceno de varios grados de reticulación con grupos SO3 -H + injertados en su superficie (intercambiadores de cationes fuertemente ácidos) o -COO -Naf (intercambiadores de cationes débilmente ácidos), -H2N + (CH3) 3Cl- (intercambiadores de aniones de base fuerte) o -N+HR2Cl- (intercambiadores de aniones de base débil).
Sorbentes para cromatografía de penetración en gel. El tipo principal es estireno-DVB. También se utilizan vidrios macroporosos, metacrilato de metilo, gel de sílice. Para la cromatografía de exclusión iónica, se utilizan los mismos adsorbentes.
Zapatillas. Para asegurar el flujo de la fase móvil (MP) a través de la columna con los parámetros especificados, se utilizan bombas de alta presión. Las especificaciones más importantes para las bombas LC son: rango de flujo; presión máxima de trabajo; reproducibilidad del flujo; rango de pulsaciones de suministro de disolvente.
Por la naturaleza del suministro de solvente, las bombas pueden ser de suministro constante (caudal) y presión constante. Básicamente, en el trabajo analítico, se usa un modo de flujo constante, cuando se llenan columnas, se usa un modo de presión constante. Según el principio de funcionamiento, las bombas se dividen en bombas de jeringa y bombas de émbolo alternativo.
bombas de jeringa Las bombas de este tipo se caracterizan por la ausencia casi total de pulsaciones en el flujo de la fase móvil durante el funcionamiento. Desventajas de la bomba: a) alto consumo de tiempo y solvente para el lavado al cambiar el solvente; b) suspensión de la separación durante el llenado de la bomba; c) grandes dimensiones y peso al mismo tiempo que proporciona un alto caudal y presión (se necesita un motor potente y un pistón de gran fuerza con su gran área).
Bombas alternativas de émbolo. Las bombas de este tipo proporcionan un caudal volumétrico constante de la fase móvil durante mucho tiempo. Presión máxima de trabajo 300-500 atm, caudal 0,01-10 ml/min. Reproducibilidad de la alimentación volumétrica - 0,5%. El principal inconveniente es que el disolvente se alimenta al sistema en forma de una serie de pulsos sucesivos, por lo que hay pulsaciones de presión y flujo.
Esta es la razón principal del aumento del ruido y la desensibilización de casi todos los detectores utilizados en LC, especialmente los electroquímicos. Formas de combatir las pulsaciones: usando bombas dobles o una bomba Bag-lai de dos émbolos, usando dispositivos de amortiguación y dispositivos electrónicos.
La cantidad de alimentación volumétrica está determinada por tres parámetros: diámetro del émbolo (generalmente 3,13; 5,0; 7,0 mm), su amplitud (12-18 mm) y frecuencia (que depende de la velocidad de rotación del motor y la caja de cambios).
Dosificadores. El propósito del dosificador es transferir una muestra a presión atmosférica a la entrada de una columna a presiones de hasta varias atmósferas. Es importante que el dispensador esté libre de volúmenes "muertos" que no puedan ser lavados por la fase móvil y que la muestra no se lave durante la dispensación. Al principio, los dispensadores en LC eran similares a los dispensadores de gas con una membrana perforada. Sin embargo, las membranas no aguantan más de 50-100 atm, su resistencia química es insuficiente, sus piezas contaminan los filtros de la columna y los capilares.
En la fase líquida, la velocidad de difusión es mucho menor que en la fase gaseosa. Por lo tanto, es posible prescindir de una parada de flujo: la muestra no tiene tiempo de desdibujarse en el dispensador. Durante la inyección de la muestra en el dispensador, un grifo especial cierra el flujo del solvente. La presión a la entrada de la columna disminuye rápidamente, después de unos segundos la muestra se puede inyectar en la cámara de dosificación con una microjeringa convencional. A continuación, se bloquea el dispensador, se activa el flujo de disolvente y tiene lugar la separación.
La presión que soporta esta válvula es de hasta 500-800 atm. Pero cuando el flujo se detiene, se altera el equilibrio en la columna, lo que puede provocar la aparición de picos adicionales "vacíos".
Los dispensadores de bucle son los más utilizados. Cuando se llena el dispensador a alta presión, hay entradas 1, 2 y un canal entre ellas. Las entradas 3-6, los canales entre ellas y el circuito de dosificación están a presión atmosférica, lo que le permite llenar el circuito con una jeringa o bomba. A medida que gira el dispensador, el flujo de la fase móvil empuja la muestra hacia la columna. Para reducir el error, el bucle se lava con 5 a 10 veces el volumen de la muestra. Si la muestra es pequeña, puede inyectarse en el asa con una microjeringa. El volumen del bucle suele ser de 5 a 50 μl.
SOBRE EL. Voinov, T. G. Volova
Cromatografía líquida Este es un método para separar y analizar mezclas complejas de sustancias en las que el líquido es la fase móvil. Es aplicable a la separación de una gama más amplia de sustancias que el método de cromatografía de gases. Esto se debe al hecho de que la mayoría de las sustancias no son volátiles, muchas de ellas son inestables a altas temperaturas (especialmente los compuestos de alto peso molecular) y se descomponen cuando pasan a estado gaseoso. La separación de sustancias por cromatografía líquida se lleva a cabo con mayor frecuencia a temperatura ambiente.
Las características de todos los tipos de cromatografía líquida se deben al hecho de que la fase móvil es un líquido, y la sorción de componentes de un eluyente gaseoso y líquido se produce de manera diferente. Si en la cromatografía de gases el gas portador realiza solo una función de transporte y no es absorbido por la fase estacionaria, entonces la fase móvil líquida en la cromatografía líquida es un eluyente activo, sus moléculas pueden ser absorbidas por la fase estacionaria. Al pasar por la columna, las moléculas de los componentes de la mezcla analizada que se encuentran en el eluyente deben desplazar las moléculas del eluyente de la capa superficial del sorbente, lo que conduce a una disminución de la energía de interacción entre las moléculas del sorbente. sustancia analizada y la superficie del adsorbente. Por lo tanto, los valores de los volúmenes retenidos V R, proporcional al cambio en la energía libre del sistema, es menor en la cromatografía líquida que en la cromatografía de gases, y el rango de linealidad de la isoterma de sorción en la cromatografía líquida es más amplio.
Mediante el uso de varios eluyentes, se pueden cambiar los parámetros de retención y la selectividad del sistema cromatográfico. La selectividad en la cromatografía líquida, a diferencia de la cromatografía de gases, está determinada no por uno, sino por dos factores: la naturaleza de las fases móvil (eluyente) y estacionaria.
La fase móvil líquida tiene mayor densidad y viscosidad que la fase gaseosa, coeficientes de difusión D bien 3-4 órdenes de magnitud menor que en el gas. Esto conduce a una ralentización de la transferencia de masa en la cromatografía líquida en comparación con la cromatografía de gases. La ecuación de Van Deemter debido al hecho de que el término V no juega un papel en la cromatografía líquida ( D bien D GRAMO), la dependencia gráfica de la eficiencia también cambia H sobre la velocidad lineal del flujo de la fase móvil tiene la forma que se muestra en la fig. 1.9. 

En la versión clásica de la cromatografía líquida en columna, se inyecta la muestra analizada disuelta en el eluyente en una columna de vidrio de 1 a 2 m de altura, llena con un sorbente con un tamaño de partícula de 100 μm y eluyente, y el eluyente se pasa a través de , tomando porciones del eluato a la salida de la columna. Esta versión de la cromatografía líquida todavía se usa en la práctica de laboratorio, pero dado que la velocidad de flujo del eluyente bajo la acción de la gravedad es baja, el análisis es largo.
La versión moderna de la cromatografía líquida, la denominada HPLC de cromatografía líquida de alto rendimiento, utiliza sorbentes volumétricos y porosos en la superficie con un tamaño de partícula de 5 a 10 μm, bombas de presión que proporcionan una presión en el sistema de hasta 400 atm y altamente detectores sensibles. La transferencia de masa rápida y la alta eficiencia de separación hacen posible el uso de HPLC para la separación de moléculas (cromatografía de adsorción de líquido y partición líquido-líquido), para la separación de iones (cromatografía de intercambio de iones, iones, pares de iones), para la separación de macromoléculas (cromatografía de exclusión por tamaño).
1.3. RETENCIÓN Y FUERZA DEL SOLVENTE
Para que los analitos se separen en la columna, como se mencionó anteriormente, el factor de capacidad k" debe ser mayor que 0, es decir, las sustancias deben ser retenidas por la fase estacionaria, el adsorbente. Sin embargo, el factor de capacidad no debe ser demasiado grande para obtener un tiempo de elución aceptable.Si se elige una fase estacionaria para una mezcla dada de sustancias, que las retiene, entonces el trabajo adicional en el desarrollo de un procedimiento de análisis consiste en elegir un solvente que proporcione, en el caso ideal, diferentes para todos los componentes, pero aceptable k no muy grande. Esto se logra cambiando la fuerza de elución del solvente.

En el caso de la cromatografía de adsorción en gel de sílice o alúmina, por regla general, la fuerza de un solvente de dos componentes (por ejemplo, hexano con la adición de isopropanol) aumenta al aumentar el contenido del componente polar (isopropanol) en él. , o reducido al disminuir el contenido de isopropanol. Si el contenido del componente polar se vuelve demasiado bajo (menos del 0,1 %), debe reemplazarse con una fuerza de elución más débil. Lo mismo se hace reemplazando el componente polar o no polar por otros, aunque este sistema no proporcione la selectividad deseada con respecto a los componentes de la mezcla de interés. A la hora de seleccionar los sistemas de disolventes se tienen en cuenta tanto las solubilidades de los componentes de la mezcla como las series eluotrópicas de disolventes recopiladas por diferentes autores.
Aproximadamente de la misma manera, la fuerza del solvente se selecciona en el caso de usar fases polares injertadas (nitrilo, amino, diol, nitro, etc.), teniendo en cuenta posibles reacciones químicas y excluyendo solventes peligrosos para la fase (por ejemplo , aldehídos y cetonas para la fase amino).
En el caso de la cromatografía de fase inversa, la fuerza del disolvente aumenta aumentando el contenido del componente orgánico (metanol, acetonitrilo o THF) en el eluyente y se reduce añadiendo más agua. Si no se puede lograr la selectividad deseada, se usa otro componente orgánico o se intenta cambiarlo con la ayuda de varios aditivos (ácidos, reactivos de par iónico, etc.).
En las separaciones por cromatografía de intercambio iónico, la fuerza del solvente se cambia aumentando o disminuyendo la concentración de la solución tampón o cambiando el pH, en algunos casos se usa la modificación con sustancias orgánicas.
Sin embargo, especialmente en el caso de mezclas biológicas y naturales complejas, a menudo no es posible elegir la concentración del solvente de tal manera que todos los componentes de la muestra sean eluidos dentro de un tiempo aceptable. Entonces uno tiene que recurrir a la elución en gradiente, es decir use un solvente cuya fuerza de elución cambie durante el análisis para que aumente constantemente de acuerdo con un programa predeterminado. Con esta técnica, es posible lograr la elución de todos los componentes de mezclas complejas en un período de tiempo relativamente corto y su separación en componentes en forma de picos estrechos.
1.6.1. Cromatografía líquida de adsorción. La cromatografía líquida de adsorción, dependiendo de la polaridad de las fases estacionaria y móvil, se divide en cromatografía de fase normal (NPC) y de fase inversa (RPC). En NPC, se utilizan un adsorbente polar y fases móviles no polares; en OFC, se utilizan un adsorbente no polar y fases móviles polares, pero en ambos casos, la elección de la fase móvil suele ser más importante que la elección del uno estacionario. La fase estacionaria debe contener las sustancias a separar. La fase móvil, es decir, el solvente, debe proporcionar diferentes capacidades de columna y separaciones eficientes en un tiempo razonable.

Como fase estacionaria en la cromatografía líquida de adsorción, se utilizan materiales porosos polares y no polares finamente dispersos con un área superficial específica de más de 50 m 2 /g. Los adsorbentes polares (SiO 2 ,Al 2 O 3 , florisil, etc.) tienen grupos débilmente ácidos en la superficie, capaces de retener sustancias con propiedades básicas. Estos adsorbentes se utilizan principalmente para la separación de compuestos no polares y polares medios. Su inconveniente es la alta sensibilidad al contenido de agua en los eluyentes utilizados. Para eliminar este inconveniente, los sorbentes polares se tratan con aminas, dioles y otros reactivos, lo que da como resultado el injerto superficial de estos reactivos, la modificación de la superficie y un cambio en la selectividad con respecto a los analitos.
Los adsorbentes no polares (hollín grafitado, tierra de diatomeas, tierra de diatomeas) no son selectivos para las moléculas polares. Para obtener adsorbentes no polares, los grupos no polares a menudo se injertan en la superficie, por ejemplo, de gel de sílice, por ejemplo, alquilsililo - SiR 3, donde los grupos alquilo R son C 2 - C 22.
La fase móvil debe disolver completamente la muestra analizada, tener una baja viscosidad (para que los coeficientes de difusión sean lo suficientemente grandes), es deseable que sea posible separar los componentes separados de ella. Debe ser inerte a los materiales de todas las partes del cromatógrafo, seguro, barato, adecuado para el detector.
Las fases móviles utilizadas en cromatografía líquida difieren en su fuerza de elución. El poder de elución del solvente muestra cuántas veces la energía de sorción de un eluyente dado en un adsorbente dado es mayor que la energía de sorción del eluyente elegido como estándar, por ejemplo, n-heptano. Los disolventes débiles son mal adsorbidos por la fase estacionaria, por lo que los coeficientes de distribución de las sustancias adsorbidas (sorbato) son altos. Por el contrario, los solventes fuertes se adsorben bien, por lo que los coeficientes de partición de sorbato son bajos. Cuanto más fuerte es el solvente, mayor es la solubilidad de la muestra analizada en él, más fuerte es la interacción solvente-sorbato.
Para garantizar una alta eficiencia de separación en la columna, es necesario elegir una fase móvil que tenga la polaridad óptima para que la mezcla se separe en las condiciones de separación seleccionadas. Normalmente, se selecciona primero la fase estacionaria, que tiene una polaridad cercana a la de los componentes a separar. Luego se selecciona la fase móvil, asegurando que el coeficiente de capacitancia k" resultó estar en el rango de 2 a 5. Si la polaridad de la fase móvil está demasiado cerca de la polaridad de la fase estacionaria, el tiempo de retención de los componentes será demasiado corto, y si las polaridades de la fase móvil y estacionaria las fases difieren mucho, el tiempo de retención se hace demasiado largo.
A la hora de seleccionar las fases móviles, se guían por las denominadas series eluotrópicas, basadas en el uso de índices de polaridad Snider R", que subdivide todos los disolventes en fuertes (polares) y débiles (débilmente polares y no polares). La escala de polaridad se basa en la solubilidad de las sustancias utilizadas como fases móviles en dioxano, nitrometano y etanol.
La tabla 1.2 muestra los valores del índice de polaridad y la fuerza de elución (con respecto al SiO 2 ) de una serie de disolventes más utilizados en cromatografía líquida como fases móviles. Los límites de transparencia de longitud de onda corta de estos solventes también se indican aquí, lo que facilita la selección de condiciones para detectar componentes de la mezcla.
Tabla 1.2
Características de los disolventes utilizados en cromatografía líquida
|
Solvente |
Índice de polaridad |
Poder de elución (SiO 2 ) |
Límite de transparencia de onda corta |
|
Fluoralcano | |||
|
ciclohexano | |||
|
norte-Hexano | |||
|
Tetracloruro de carbono | |||
|
Éter diisopropílico | |||
|
éter dietílico | |||
|
diclorometano | |||
|
tetrahidrofurano | |||
|
Cloroformo | |||
|
Ácido acético | |||
|
acetonitrilo | |||
|
nitrometano | |||
La cromatografía líquida a menudo no utiliza disolventes individuales, sino sus mezclas. A menudo, las adiciones menores de otro solvente, especialmente agua, aumentan en gran medida la fuerza de elución del eluyente.
Al separar mezclas de varios componentes, es posible que una fase móvil como eluyente no separe todos los componentes de la muestra en un tiempo razonable. En este caso, se utiliza un método de elución por pasos o en gradiente, en el que se utilizan secuencialmente eluyentes cada vez más fuertes durante la cromatografía, lo que permite eluir sustancias muy retenidas en menos tiempo.
En la cromatografía líquida, hay algunos empírico reglas que son muy útiles a la hora de elegir un eluyente:
la sorción de un compuesto, por regla general, aumenta con un aumento en el número de dobles enlaces y grupos OH en él;
la sorción disminuye en varios compuestos orgánicos: ácidosalcoholesaldehídoscetonaséstereshidrocarburos insaturadoshidrocarburos saturados;
para separar sustancias de diferente polaridad y separar sustancias de diferentes clases, se utiliza la cromatografía de fase normal: la muestra analizada se disuelve y eluye con un eluyente no polar, los compuestos de diferentes clases salen de la columna con un adsorbente polar durante diferentes tiempos, mientras que el tiempo de retención de compuestos con diferentes grupos funcionales aumenta durante la transición de compuestos no polares a compuestos débilmente polares. Para moléculas muy polares, los tiempos de retención son tan largos que el análisis no es posible cuando se utiliza un eluyente no polar. Para reducir el tiempo de retención de los componentes polares, se pasa a eluyentes polares;
en la variante de fase inversa, la fase estacionaria (adsorbente no polar) adsorbe más fuertemente el componente no polar de los eluyentes polares, por ejemplo, del agua;
Reduciendo la polaridad del eluyente añadiendo un disolvente menos polar, se puede reducir la retención de los componentes.
1.6.2. Cromatografía líquida de partición. En la cromatografía de partición o líquido-líquido, la separación de los componentes de la muestra analizada se debe a diferencias en los coeficientes de su distribución entre dos fases líquidas que no se mezclan entre sí, una de las cuales es estacionaria y se encuentra en la superficie. o en los poros de un soporte inmóvil sólido, y el segundo es móvil.
En cuanto a la naturaleza de las fuerzas de interacción que provocan una distribución diferente entre las dos fases de sustancias que difieren en su estructura química, la cromatografía de distribución es similar a la cromatografía de adsorción, es decir, aquí la separación también se basa en la diferencia en las fuerzas de interacción intermolecular entre los componentes de la muestra analizada con las fases líquidas estacionaria y móvil.
Dependiendo de la técnica de ejecución, la cromatografía de partición, como la cromatografía de adsorción, puede ser de columna o plana (papel o capa delgada).
Como portadores sólidos se utilizan sustancias indiferentes al disolvente móvil ya los componentes de la muestra analizada, pero capaces de retener la fase estacionaria en la superficie y en los poros. En la mayoría de los casos, las sustancias polares (celulosa, gel de sílice, almidón) se utilizan como vehículos. Se aplican a la fase estacionaria: un solvente polar, generalmente agua o alcohol. En este caso, se utilizan como fases móviles sustancias menos polares o no polares (alcoholes, aminas, cetonas, hidrocarburos, etc.). Este tipo de cromatografía de partición se llama fase normal. Se utiliza para separar sustancias polares.
La segunda variante de la cromatografía de partición se diferencia en que se utilizan portadores no polares (goma, PTFE, gel de sílice hidrofobizado) como fase sólida estacionaria, disolventes no polares (hidrocarburos) como fase líquida estacionaria y disolventes polares (alcoholes, aldehídos ) como la fase líquida móvil, cetonas, etc., a menudo agua). Esta variante de la cromatografía de partición se llama fase inversa y se utiliza para separar sustancias no polares.
Para lograr una separación óptima de los componentes de la muestra analizada, la selección de la fase móvil es muy importante. Los solventes (fases líquidas móviles y estacionarias) deben seleccionarse de modo que los coeficientes de distribución de los componentes de la mezcla difieran significativamente. Las fases líquidas están sujetas a las siguientes requisitos:
1) los disolventes utilizados deben disolver bien las sustancias a separar, y su solubilidad en la fase estacionaria debe ser mayor que en la móvil;
2) los disolventes utilizados como fase móvil y estacionaria deben estar mutuamente saturados, es decir, la composición del disolvente debe ser constante durante el paso por la columna;
3) la interacción de los disolventes utilizados como fase móvil con la fase estacionaria debe ser mínima.

Muy a menudo, en la cromatografía líquida de partición, no se utilizan sustancias individuales como fases líquidas móviles, sino sus mezclas en varias proporciones. Esto permite regular la polaridad de la fase móvil, cambiar la relación de las polaridades de las fases móvil y estacionaria y lograr condiciones óptimas para separar los componentes de una mezcla particular que se analiza.
Una desventaja significativa de este método cromatográfico es el lavado bastante rápido de la fase líquida estacionaria depositada del soporte. Para eliminarlo, el disolvente utilizado como fase móvil se satura con la sustancia utilizada como fase líquida estacionaria, o se estabiliza la fase líquida estacionaria injertándola sobre un soporte.
Una variación de la cromatografía líquida de partición es el método HPLC ampliamente utilizado.

Los sistemas cromatográficos más comunes son sistemas que tienen un principio de montaje modular. Las bombas, los desgasificadores, los detectores, los dispensadores (automuestreadores), los hornos de columna, los colectores de fracciones, los controles del sistema de cromatografía y los registradores están disponibles como módulos separados. Una amplia gama de módulos le permite resolver de manera flexible varios problemas analíticos, cambiar rápidamente la configuración del sistema si es necesario, con costos mínimos. Al mismo tiempo, también se producen LC monomodulares (integrados), cuya principal ventaja es la miniaturización de bloques individuales y la compacidad del dispositivo.
Según el método de elución, los cromatógrafos líquidos se dividen en isocráticos y de gradiente.
Diagrama de un cromatógrafo isocrático
La fase móvil desde el tanque (1) a través del filtro de entrada (9) es suministrada por una bomba de precisión de alta presión (2) al sistema de entrada de muestra (3) - un inyector manual o un automuestreador, donde también se inyecta la muestra . Además, a través del filtro en línea (8), la muestra con la corriente de la fase móvil ingresa al elemento de separación (elementos) (4), a través de la precolumna a la columna de separación. Luego, el eluato ingresa al detector (5) y se retira al tanque de drenaje (7). Cuando el eluato fluye a través del circuito de medición del detector, el cromatograma se registra y los datos se transmiten a un registrador analógico (registrador) (6) u otro sistema para recolectar y procesar datos cromatográficos (integrador o computadora). Dependiendo del diseño de los módulos funcionales, el sistema puede controlarse desde el teclado del módulo de control (generalmente un controlador de bomba o sistema), desde los teclados de cada uno de los módulos del sistema, o mediante un programa de control desde una computadora personal.
En el caso de la elución en gradiente, se utilizan dos tipos fundamentalmente diferentes de cromatógrafos líquidos. Se diferencian en el punto de formación del gradiente de composición de la fase móvil.
Esquema de un cromatógrafo de gradiente con la formación de un gradiente de la composición de la fase móvil en la línea de baja presión.
La fase móvil desde los tanques (1) a través de los filtros de entrada (9) y el programador de gradientes (10) es suministrada por una bomba de precisión de alta presión (2) al sistema de inyección de muestra (3) - un inyector manual o un automuestreador , donde también se inyecta la muestra. El funcionamiento de las válvulas del programador de gradiente está controlado por el módulo de control del sistema (bomba o controlador) o por un programa de control de PC. Los sistemas de este tipo forman un gradiente binario, tridimensional y tetradimensional. La forma de la función de procesamiento de gradiente depende del módulo de control o programa de control específico, así como de la funcionalidad de los módulos controlados y de control. Además, a través del filtro en línea (8), la muestra con la corriente de la fase móvil ingresa al elemento de separación (elementos) (4), a través de la precolumna a la columna de separación. Luego, el eluato ingresa al detector (5) y se retira al tanque de drenaje (7). Cuando el eluato fluye a través del circuito de medición del detector, el cromatograma se registra y los datos se transmiten a un registrador analógico (registrador) (6) u otro sistema para recolectar y procesar datos cromatográficos (integrador o computadora). Dependiendo del diseño de los módulos funcionales, el sistema puede controlarse desde el teclado del módulo de control (generalmente un controlador de bomba o sistema) o mediante un programa de control desde una computadora personal. En el caso de controlar el módulo de control, es posible controlar el detector de forma independiente desde su propio teclado.
A pesar del aparente atractivo de tales sistemas (utilizan sólo una bomba de precisión de alta presión), estos sistemas tienen una serie de desventajas, entre las cuales la principal, quizás, es la severa necesidad de una desgasificación completa de los componentes de la fase móvil incluso antes de la mezclador de baja presión (cámara del programador de gradientes). Se lleva a cabo con la ayuda de desgasificadores de flujo especiales. Debido a este hecho, su costo se vuelve comparable a otro tipo de sistemas de gradiente: sistemas con la formación de una composición de gradiente de la fase móvil en la línea de alta presión.
La diferencia fundamental entre los sistemas con la formación de una composición de gradiente de fase móvil en la línea de alta presión es la mezcla de componentes en la línea de alta presión, naturalmente, con este enfoque, el número de bombas de precisión está determinado por el número de tanques para mezclar. la fase móvil. Con este enfoque, los requisitos para la minuciosidad de la desgasificación de los componentes se reducen significativamente.
Esquema de un cromatógrafo de gradiente con la formación de un gradiente de la composición de la fase móvil en la línea de alta presión.
La fase móvil desde los tanques (1) a través de los filtros de entrada (9) es suministrada por bombas de alta presión de precisión (2 y 11) a través de un mezclador de flujo estático o dinámico (10) al sistema de inyección de muestra (3) - un manual inyector o un automuestreador, donde también se inyecta la muestra. El funcionamiento de las bombas controladas está controlado por el módulo de control del sistema (bomba maestra o controlador) o por un programa de control de PC. En este caso, todas las bombas están controladas. Los sistemas de este tipo forman un gradiente binario o tridimensional. La forma de la función de procesamiento de gradiente depende del módulo de control o programa de control específico, así como de la funcionalidad de los módulos controlados y de control. Además, a través del filtro en línea (8), la muestra con la corriente de la fase móvil ingresa al elemento de separación (elementos) (4), a través de la precolumna a la columna de separación. Luego, el eluato ingresa al detector (5) y se retira al tanque de drenaje (7). Cuando el eluato fluye a través del circuito de medición del detector, el cromatograma se registra y los datos se transmiten a un registrador analógico (registrador) (6) u otro sistema para recolectar y procesar datos cromatográficos (integrador o computadora). Dependiendo del diseño de los módulos funcionales, el sistema puede controlarse desde el teclado del módulo de control (generalmente un controlador de bomba o sistema) o mediante un programa de control desde una computadora personal. En el caso de controlar el módulo de control, es posible controlar el detector de forma independiente desde su propio teclado.
Los esquemas propuestos están bastante simplificados. Se pueden incluir dispositivos adicionales en los sistemas: un termostato de columna, sistemas de derivatización posterior a la columna, sistemas de preparación y concentración de muestras, un reciclador de solventes, sistemas de membrana para suprimir la conductividad eléctrica de fondo (para cromatografía iónica), sistemas de protección adicionales (filtros, columnas) , etc En los diagramas, los módulos manométricos tampoco se muestran por separado. Como regla general, estos dispositivos están integrados en unidades de bomba. Estas unidades pueden combinar varias bombas, una bomba con un programador de gradiente y un controlador de sistema común. La estructura del sistema depende de su configuración y de cada fabricante en concreto.
Una complicación tan radical del soporte técnico del proceso cromatográfico conduce a la aparición de una serie de requisitos para las propiedades de la fase móvil, que están ausentes en la cromatografía clásica de columna y plana. La fase líquida debe ser apta para la detección (transparente en una determinada región del espectro o tener un índice de refracción bajo, cierta conductividad o permitividad eléctrica, etc.), inerte a los materiales de las partes del tracto cromatográfico, no formar burbujas de gas en las válvulas de la bomba y la celda del detector, no tener impurezas mecánicas.

En cromatografía líquida se utilizan muchos tipos de bombas. LC de baja presión a menudo utiliza bombas peristálticas (Figura 1).
Fig.1 Bomba peristáltica programable MasterFlex.
En HPLC se utilizan bombas de alta presión para asegurar el flujo de la fase móvil a través de la columna con los parámetros especificados.
Las características técnicas más importantes de las bombas HPLC son: rango de flujo; presión máxima de trabajo; reproducibilidad del flujo; rango de pulsaciones de suministro de disolvente.
Por la naturaleza del suministro de solvente, las bombas pueden ser de suministro constante (caudal) y presión constante. Básicamente, en el trabajo analítico, se usa un modo de flujo constante, cuando se llenan columnas, se usa un modo de presión constante.
Según el principio de funcionamiento, las bombas HPLC se dividen en jeringuilla y en émbolo recíproco .
Bombas de jeringa
La principal característica distintiva de estas bombas es su funcionamiento cíclico y, por lo tanto, los cromatógrafos en los que se utilizan estas bombas también difieren en el funcionamiento cíclico.
Arroz. 2. Disposición principal de una bomba de jeringa para HPLC.
Arroz. 2A. Bomba de jeringa.
La unidad de control BU suministra tensión al motor D, que determina la velocidad y la dirección de su rotación. La rotación del motor con la ayuda de la caja de cambios P se convierte en el movimiento del pistón P dentro del cilindro D. La bomba funciona en 2 ciclos. En el ciclo de llenado, la válvula K2 está cerrada, K1 está abierta, el solvente fluye desde el depósito al cilindro C. En el modo de suministro, la válvula K1 está cerrada y, a través de la válvula K2, la fase móvil ingresa al dispositivo de dosificación.
Las bombas de este tipo se caracterizan por la ausencia casi total de pulsaciones en el flujo de la fase móvil durante el funcionamiento.
Desventajas de la bomba:
a) alto consumo de tiempo y solvente para el lavado al cambiar el solvente;
b) el volumen de PF limitado por el volumen de la jeringa y, por lo tanto, el tiempo de separación limitado;
c) suspensión de la separación durante el llenado de la bomba;
d) grandes dimensiones y peso al mismo tiempo que proporciona un alto caudal y presión (se necesita un motor potente y un pistón de gran fuerza con su gran área).
Bombas alternativas de émbolo.
Arroz. 3. Dispositivo principal de una bomba de émbolo.
Principio de operación.
El motor D a través de la caja de cambios R mueve alternativamente el émbolo P, moviéndose en el cabezal de trabajo de la bomba. Las válvulas K1 y K2 se abren cuando la bomba está en fase de aspiración e impulsión respectivamente. La cantidad de alimentación volumétrica está determinada por tres parámetros: diámetro del émbolo (generalmente 3,13; 5,0; 7,0 mm), su amplitud (12-18 mm) y frecuencia (que depende de la velocidad de rotación del motor y la caja de cambios).

Las bombas de este tipo proporcionan un caudal volumétrico constante de la fase móvil durante mucho tiempo. Presión máxima de trabajo 300-500 atm, caudal 0,01-10 ml/min. Repetibilidad de alimentación por volumen -0,5%. El principal inconveniente es que el disolvente se introduce en el sistema en forma de una serie de pulsos sucesivos, por lo que hay pulsaciones de presión y flujo (Fig. 4). Esta es la razón principal del aumento del ruido y la desensibilización de casi todos los detectores utilizados en LC, especialmente los electroquímicos.
Figura 4. Pulsación de la bomba de émbolo.
Formas de lidiar con las pulsaciones.
1. Aplicación de dispositivos de amortiguación..
Son tubos en espiral de perfil especial fabricados en acero inoxidable, incluidos en serie o en paralelo en el sistema entre la bomba y el dosificador.
Arroz. 5. Amortiguador espiral.
El amortiguador se desenrosca con un aumento de presión en él (aceleración de la bomba). Cuando baja la presión, gira, disminuye su volumen, exprime parte del disolvente, manteniendo un caudal constante y reduciendo las pulsaciones. Tal amortiguador funciona bien a una presión de 50 atm y superior.
A una presión de 5-30 atm suaviza mejor las pulsaciones amortiguador de aire, hecho de una columna (Fig. 6.). El aire de la columna taponada (6x200 mm) se comprime y se extinguen las pulsaciones. El aire que contiene se disuelve en 24 horas.
Arroz. 6. Compuerta de aire.
2. El uso de dispositivos electrónicos.
Cuando se usa un transductor de presión electrónico, las lecturas del transductor se pueden usar para controlar el funcionamiento de la bomba. Cuando la presión cae, la velocidad del motor aumenta y compensa la disminución de la presión. También es posible compensar las fugas en las válvulas y parcialmente en los manguitos. El uso de un amortiguador electrónico (BPZh-80, KhPZh-1, etc.) permite reducir las pulsaciones de presión a 1 atm a una presión de 100-150 kgf/cm2.
1.6.3. Cromatografía de intercambio iónico, ion, par iónico. Los métodos de cromatografía de intercambio iónico, de iones y de pares de iones se basan en el proceso dinámico de sustitución de iones asociados con la fase estacionaria con iones eluyentes que ingresan a la columna. El objetivo principal del proceso cromatográfico es la separación de iones inorgánicos u orgánicos del mismo signo. La retención en estos tipos de cromatografía está determinada por el cambio en la energía libre de la reacción de intercambio iónico. La relación de las concentraciones de intercambio de iones en la solución y en la fase sorbente se caracteriza por el equilibrio de intercambio iónico. El intercambio iónico consiste en el hecho de que algunas sustancias (intercambiadores de iones), cuando se sumergen en una solución electrolítica, absorben cationes o aniones de ella, liberando una cantidad equivalente de otros iones con una carga del mismo signo en la solución. Entre el intercambiador de cationes y la solución hay un intercambio de cationes, entre el intercambiador de aniones y la solución hay un intercambio de aniones.
Los intercambiadores de cationes suelen ser sustancias poliméricas insolubles especialmente sintetizadas que contienen grupos ionogénicos ácidos en su estructura: -SO 3 H; –COOH; -OH; –PO3H2; –AsO3H2.
Las fórmulas químicas de los intercambiadores de cationes se pueden representar esquemáticamente como R-SO 3 H; R-SO 3 Na. En el primer caso, el intercambiador de cationes está en forma de H, en el segundo, en forma de Na. R es una matriz polimérica.
Las reacciones de intercambio catiónico se escriben como reacciones químicas heterogéneas ordinarias:
RN + Na + RNa + H +
Los intercambiadores de aniones contienen grupos ionogénicos básicos en su estructura: –N(CH 3) 3 + ; =NH 2 + ; =NH + etc. Sus fórmulas químicas se pueden representar como RNH 3 OH y RNH 3 Cl o ROH, RCl. En el primer caso, el intercambiador de aniones está en forma de OH, en el segundo, en forma de Cl. La reacción de intercambio aniónico se puede escribir de la siguiente manera:
R–OH+Cl – RCl+OH –
Se conocen intercambiadores de iones anfóteros que contienen grupos tanto ácidos como básicos en su estructura. Los intercambiadores de iones que tienen en su composición grupos ácidos (básicos) del mismo tipo (por ejemplo, SO 3 H) se denominan monofuncionales; intercambiadores de iones que contienen heterogéneos (por ejemplo, - SO 3 H, - OH) grupos ácidos (básicos) - polifuncionales.
Los intercambiadores de iones monofuncionales se obtienen mediante una reacción de polimerización. La reacción de policondensación permite obtener intercambiadores de iones polifuncionales. Para que los intercambiadores de iones resultantes tengan características de rendimiento suficientemente altas, deben ser insolubles, pero hinchables en el disolvente apropiado y tener un número suficientemente grande de grupos ionogénicos capaces de intercambiarse con los grupos ionogénicos de la muestra analizada. Esto se puede lograr si las cadenas de polímero resultantes están suficientemente ramificadas y conectadas entre sí mediante "puentes de reticulación". Por ejemplo, en la preparación de intercambiadores de cationes de tipo polimerización a base de estireno, el divinilbenceno se usa con mayor frecuencia como agente de reticulación, cuya introducción en una cantidad de hasta el 16% asegura la producción de intercambiadores de iones con varios grados de hinchamiento y, por lo tanto, permite controlar la porosidad del intercambiador de iones. El grado de hinchamiento del intercambiador de iones, expresado en mililitros/gramo, es el volumen de 1 g de intercambiador de iones secado al aire contenido en la columna.
El intercambiador de iones absorbe, por regla general, uno de los contraiones: iones en la fase móvil, es decir, exhibe una cierta selectividad. Se han establecido experimentalmente series de afinidad, o selectividad, de iones con respecto a intercambiadores de iones de varios tipos. Por ejemplo, a bajas concentraciones de solución en intercambiadores de cationes fuertemente ácidos, los iones con la misma carga se adsorben en la siguiente secuencia:
Li + Na + K + Rb + Cs +
Mg 2+ Ca 2+ Sr 2+ Ba 2+ .
Para iones con diferentes cargas, la capacidad de absorción aumenta al aumentar la carga:
Na+Ca2+ Sin embargo, cambiar las condiciones para llevar a cabo la reacción de intercambio iónico puede conducir a una inversión en serie. También se han establecido series de afinidad para intercambiadores de aniones. Por ejemplo, la absorbibilidad de aniones en intercambiadores de aniones fuertemente básicos aumenta en la serie: F - OH - Cl - Br - NO 3 - J - SCN - ClO 4 - . Los intercambiadores de iones que contienen grupos fuertemente ácidos o fuertemente básicos en su estructura entran en reacciones de intercambio iónico con cualquier ion en solución que tenga cargas del mismo signo que el signo del contraión. Tales intercambiadores de iones se llaman universales. El proceso de intercambio de iones entre el analito y el intercambiador de iones se puede llevar a cabo de tres formas: estática, dinámica (método de filtro de intercambio de iones) y cromatográfica. Método estático
El intercambio iónico consiste en que una muestra del intercambiador iónico se pone en contacto con un cierto volumen de solución y se agita o agita durante un tiempo determinado hasta que se establece el equilibrio. Este es un método rápido y simple de intercambio de iones, que se utiliza para concentrar iones de soluciones diluidas, eliminar impurezas no deseadas, pero no proporciona una absorción completa de iones, ya que el intercambio de iones es un proceso de no equilibrio y, por lo tanto, no garantiza una separación completa. de iones Al realizar el intercambio iónico de forma dinámica
se pasa una solución a través de la columna con un intercambiador de iones que, a medida que se mueve a lo largo de la columna, entra en contacto con nuevos gránulos del intercambiador de iones. Este proceso proporciona un intercambio más completo que el método estático, ya que los productos de intercambio son eliminados por el flujo de la solución. Pueden concentrar iones de soluciones diluidas y separar iones que difieren mucho en propiedades, por ejemplo, iones con diferente carga (separar cationes de aniones), pero la separación de iones del mismo signo de carga es casi imposible. La separación cuantitativa de dichos iones solo es posible con la repetición repetida de actos elementales de sorción-desorción en condiciones dinámicas, es decir, método cromatográfico
. Cuando se trabaja con este método, se utilizan capas altas de intercambiador de iones, y la mezcla a separar se introduce en esta capa en una cantidad mucho menor que la capacidad de la columna, por lo que se asegura la repetición repetida de actos elementales de intercambio de iones. . En cuanto a la técnica de análisis, la cromatografía de intercambio iónico es similar a la cromatografía molecular y se puede realizar según las opciones de eluyente (revelado), frontal y de desplazamiento. La diferencia entre la cromatografía molecular y la de intercambio iónico es que, en la cromatografía molecular, los componentes separados de la mezcla se eluyen de la columna con un eluyente puro, mientras que en la cromatografía de intercambio iónico se usa una solución electrolítica como eluyente. En este caso, el ión intercambiado del eluyente debe ser absorbido de manera menos selectiva que cualquiera de los iones de la mezcla que se está separando. Cuando se desarrolla la cromatografía de intercambio iónico, que se usa con más frecuencia, una columna llena con un intercambiador de iones se lava primero con una solución electrolítica hasta que el intercambiador de iones reemplaza completamente todos sus iones con los iones contenidos en el eluyente. Luego, se inyecta en la columna un pequeño volumen de la solución de analito que contiene iones separables en una cantidad de aproximadamente el 1% de la capacidad del intercambiador de iones. A continuación, la columna se lava con una solución eluyente, tomando fracciones del eluato y analizándolas. Una mezcla de iones Cl - , Br - , J - se puede separar en una resina de intercambio aniónico altamente básica (poliestireno reticulado que contiene grupos de bases de amonio cuaternario N (CH 3) 3 +), por ejemplo, AB-17, que tiene un rango de selectividad (selectividad): NO 3 - Cl – Br – J – . Como resultado, se utiliza una solución de NaNO 3 como eluyente. Primero, esta solución se pasa a través del intercambiador de iones hasta que esté completamente saturada con iones NO 3 -. Cuando la mezcla a separar se introduce en la columna, los iones Cl – , Br – , J – son absorbidos por el intercambiador de aniones, desplazando a los iones NO 3 –. Durante el lavado posterior de la columna con una solución de NaNO 3 , los iones Cl – , Br – , J – en las capas superiores del intercambiador de aniones se reemplazan gradualmente por iones NO 3 –. Los iones Cl - se desplazarán más rápido de todos, los iones J - permanecerán en la columna por más tiempo. La diferencia en la selectividad del intercambiador de iones a los iones de la mezcla conduce al hecho de que se forman zonas separadas de iones adsorbidos Cl - , Br - y J - en la columna, moviéndose a lo largo de la columna a diferentes velocidades. A medida que se desplaza por la columna, la distancia entre zonas aumenta. En cada zona solo hay uno de los aniones de la mezcla que se separa y el anión del eluyente, en el intervalo entre las zonas solo hay un anión del eluyente. Así, las fracciones que contienen componentes individuales de la mezcla a separar aparecerán en el eluyente a la salida de la columna. Para resolver problemas prácticos, las condiciones para la separación de iones se varían seleccionando una fase móvil adecuada (composición, concentración, pH, fuerza iónica) o cambiando la porosidad de la matriz polimérica del intercambiador de iones, es decir, el número de enlaces entre cadenas en la matriz, y creando tamices de intercambio iónico que sean permeables a algunos iones y capaces de su intercambio e impenetrables para otros. También es posible cambiar la naturaleza y la disposición mutua de los grupos ionogénicos, así como obtener adsorbentes capaces de reacciones químicas selectivas debido a la formación de complejos. Se posee una alta selectividad, por ejemplo, complejando intercambiadores de iones que contienen en su estructura grupos quelantes de reactivos orgánicos dimetilglioxima, ditizona, 8-hidroxiquinolina, etc., así como éteres corona. La mayor aplicación en cromatografía de intercambio de iones, iones y pares de iones se encuentra en los intercambiadores de iones orgánicos macro y micronet sintéticos que tienen una gran capacidad de intercambio (3–7 mmol/g), así como en los materiales de intercambio de iones inorgánicos. Los intercambiadores de iones de micromalla son capaces de intercambiar iones solo en estado hinchado, macromalla, en estados hinchados y no hinchados. Otro tipo estructural de intercambiadores de iones son los intercambiadores de iones de película superficial, cuyo núcleo sólido está hecho de un copolímero no poroso de estireno y divinilbenceno, vidrio o gel de sílice y está rodeado por una película delgada del intercambiador de iones. El diámetro total de una partícula de este tipo es de unos 40 µm y el espesor de la película de ionita es de 1 µm. La desventaja de tales intercambiadores de iones es el diámetro de partícula relativamente grande y la baja capacidad de intercambio debido a la baja área superficial específica, por lo que es necesario trabajar con muestras pequeñas y, en consecuencia, usar detectores altamente sensibles. Además, tales intercambiadores de iones se envenenan rápidamente y no son capaces de regenerarse. En intercambio iónico de alto rendimiento y cromatografía iónica, intercambiadores de iones de poliestireno poroso volumétrico, sílices porosos volumétricos con un diámetro de gránulo de aproximadamente 10 μm, y copolímeros de estireno y divinilbenceno de superficie porosa y modificados en la superficie prácticamente que no se hinchan con ionogénico Se utilizan grupos sulfo y amino. En la cromatografía de pares de iones, se utilizan adsorbentes de "cepillo": geles de sílice con fases inversas injertadas C 2, C 8, C 18, que se convierten fácilmente en un intercambiador de cationes tras la absorción de tensioactivos iónicos de la fase móvil, por ejemplo, alquilo sulfatos o sales de bases de amonio cuaternario. Cuando se lleva a cabo la separación cromatográfica utilizando intercambiadores de iones, las soluciones acuosas de sales se utilizan con mayor frecuencia como fase móvil. Esto se debe al hecho de que el agua tiene excelentes propiedades de disolución e ionización, por lo que las moléculas de la muestra analizada se disocian instantáneamente en iones, los grupos de intercambio iónico del intercambiador de iones se hidratan y también se disocian total o parcialmente. Esto asegura un rápido intercambio de contraiones. La fuerza de elución de la fase móvil está influenciada principalmente por el pH, la fuerza iónica, la naturaleza de la solución tampón, el contenido de solvente orgánico o tensioactivo (cromatografía de par iónico). El valor del pH se elige en función de la naturaleza de los grupos ionogénicos, los iones a separar y la matriz. Es posible trabajar con intercambiadores de iones fuertemente ácidos y fuertemente básicos a pH = 2–12, con débiles ácidos a pH = 5–12, y débilmente básicos a pH = 2–6. Los adsorbentes a base de sílice no se pueden usar a pH 9. La fuerza iónica de la fase móvil afecta la capacidad del intercambiador de iones. Con un aumento de la fuerza iónica, la sorción de iones suele disminuir, ya que aumenta la fuerza de elución de la fase móvil. Por lo tanto, al comienzo de la separación, la fase móvil debe tener una fuerza iónica baja (0,05–0,1), y el valor final de esta característica no debe exceder 2. En la elución en gradiente, a menudo se usan tampones con fuerza iónica creciente. Para la elución selectiva de los iones absorbidos por el intercambiador de iones, se puede utilizar agua, soluciones tampón (fosfato, acetato, borato, hidrocarbonato, etc.) con un determinado valor de pH y fuerza iónica, soluciones de minerales (clorhídrico, nitrogenado, sulfúrico, fosfórico) y orgánicos (fenol, cítrico, láctico, tartárico, oxálico, EDTA). La elección del eluyente se ve facilitada por el hecho de que los coeficientes limitantes de distribución de la mayoría de los elementos entre soluciones acuosas (agua-orgánica) de muchos complejantes e intercambiadores de iones de tipo estándar se determinan y presentan en tablas. 1.6.4. Cromatografía de exclusión por tamaño. La cromatografía de exclusión por tamaño es un tipo de cromatografía líquida en la que la separación de componentes se basa en la distribución de moléculas según su tamaño entre el disolvente en los poros del sorbente y el disolvente que fluye entre sus partículas. Durante la separación, las moléculas pequeñas ingresan a la red de polímeros, en cuyos poros el solvente sirve como fase estacionaria, y se retienen allí. Las moléculas grandes no pueden penetrar en la red de polímeros y la fase móvil las elimina de la columna. Primero se eluyen las moléculas más grandes, luego las medianas y finalmente las pequeñas. La cromatografía de exclusión por tamaño se subdivide en permeación en gel y filtración en gel. En la cromatografía de permeación en gel, la separación se produce en polímeros que se hinchan en disolventes orgánicos. La versión de filtración en gel de la cromatografía de exclusión por tamaño implica el uso de polímeros hinchables en agua como fases estacionarias. El tiempo de retención de los componentes de la muestra analizada en la columna de exclusión por tamaño depende del tamaño de sus moléculas y de la difusión en los poros del adsorbente, así como del tamaño de los poros de la fase estacionaria. En este tipo de cromatografía líquida, el coeficiente de partición D para las moléculas más pequeñas de la muestra analizada, que se mueven en la columna cromatográfica a menor velocidad, penetrando en la rejilla de la fase estacionaria, es igual a 1, ya que la fase móvil y el solvente en los poros de la fase estacionaria tienen la misma composición. En este caso, la ecuación principal de la cromatografía en columna toma la forma Las moléculas grandes que no entran en los poros de la fase estacionaria se eluyen de la columna junto con la fase móvil. Para ellos D= 0, un V R =V metro. Tal rango de valores del coeficiente de distribución (de 0 a 1) es típico solo para la cromatografía de exclusión por tamaño. Todas las moléculas de la sustancia multicomponente analizada deben eliminarse de la columna pasando un pequeño volumen de disolvente de V metro antes de V metro +V s y la separación se completa antes de que salga el pico de disolvente. Por tanto, en este tipo de cromatografía es necesario utilizar columnas suficientemente largas y con un gran volumen libre. V metro y un gran número de poros en el adsorbente. La resolución de los picos cromatográficos en las separaciones por exclusión de tamaño se puede mejorar mediante el uso de gradientes de elución con disolventes mixtos. Cada sorbente utilizado en la cromatografía de exclusión por tamaño se caracteriza por un determinado volumen de poro y, por lo tanto, tiene una determinada área de pesos moleculares separables y una determinada curva de calibración. En este caso, la curva de calibración que caracteriza la dependencia del volumen retenido del peso molecular o tamaño de las moléculas, por regla general, tiene una forma compleja. Las fases estacionarias en la cromatografía de exclusión por tamaño se seleccionan en función de tareas analíticas específicas. Inicialmente, se establece qué sistema de disolventes se puede utilizar para el análisis (acuoso o agua-orgánico). Dependiendo de esto, se determina el tipo de sorbente. Si se van a separar muestras solubles en agua, se utilizan, por ejemplo, dextranos reticulados hinchables en agua (Sephadex) o poliacrilamidas (Biogel P) como fases estacionarias. La separación de sustancias solubles en disolventes orgánicos se puede realizar sobre poliestirenos con diversos grados de reticulación, que se hinchan en disolventes orgánicos (styrogel, poragel, biobid C). Dichos geles hinchados generalmente no son estables a la presión y permiten velocidades de flujo de fase móvil muy bajas, lo que aumenta el tiempo de análisis. Para implementar una versión altamente eficiente de cromatografía de exclusión por tamaño, es necesario utilizar fases estacionarias con matrices rígidas - geles de sílice, cuya desventaja - alta actividad de adsorción - se elimina mediante la silanización de la superficie o la selección de un eluyente de la polaridad adecuada . Las sustancias que se pueden utilizar como fases móviles en la cromatografía de exclusión por tamaño son: disolver completamente la muestra analizada; humedecer bien el sorbente; contrarrestar la adsorción de los componentes de la muestra en el adsorbente; tienen baja viscosidad y toxicidad. 1.6.5. cromatografía planar. La cromatografía planar incluye la cromatografía en capa fina y en papel. Estos tipos de cromatografía líquida son de técnica simple, rápida, no requieren equipos costosos, lo cual es su innegable ventaja. La separación de una mezcla de sustancias por estos métodos se puede realizar utilizando varios sistemas cromatográficos. Por lo tanto, se distinguen adsorción, distribución, fase normal e inversa, intercambio iónico, etc., cromatografía en papel y en capa fina. En la actualidad, la cromatografía en capa fina es la más utilizada. La cromatografía en papel y en capa fina son similares en técnica. La fibra de papel de celulosa se utiliza como fase estacionaria en la cromatografía en papel, en la cromatografía en capa fina: varios sorbentes (Al 2 O 3 , gel de sílice, etc.) depositados en una capa delgada uniforme (100-300 μm) sobre un vidrio, metal o sustrato de plástico (portador). La capa adsorbente sobre el soporte puede ser fija o no. La separación cromatográfica en métodos planos, así como en columna, se debe a la transferencia de los componentes del analito por parte de la fase móvil a lo largo de la capa de la fase estacionaria a diferentes velocidades de acuerdo con los coeficientes de distribución de las sustancias a separar. . En ambos casos se utilizan sistemas cromatográficos sorbentes líquido-sólido (mecanismo de separación por adsorción), portador líquido-líquido-sólido (distribución, intercambio iónico y otros mecanismos). Como fases móviles se utilizan diversos disolventes o sus mezclas, ácidos orgánicos o inorgánicos. La práctica de obtención de cromatogramas planos consiste en lo siguiente. Sobre una tira de papel cromatográfico o sobre una fina capa de adsorbente, se marca con un lápiz una línea de partida a una distancia de 1 cm del borde inferior de la tira o placa. Se utiliza una micropipeta para aplicar una muestra en la línea de salida en forma de punto con un diámetro de no más de 2-3 mm. Luego, el borde de la tira o placa se baja al recipiente con la fase móvil, ubicada en una cámara sellada. A medida que la fase móvil asciende a lo largo de la tira o placa y se producen los múltiples actos elementales de sorción-desorción, distribución entre dos fases líquidas, intercambio iónico, etc., que son habituales en la cromatografía, se separan los componentes de la mezcla analizada. El proceso generalmente continúa hasta que el solvente ha pasado de la línea de inicio 10 cm Después de eso, la tira o placa se retira de la cámara y se seca. Si los componentes del analito están coloreados, dan las manchas de color correspondientes en el cromatograma. Para detectar componentes no teñidos del analito, se debe desarrollar el cromatograma. El desarrollo de un cromatograma y la detección de los componentes de la muestra se pueden realizar por varios métodos y dependen de la composición de las mezclas analizadas. La manifestación se puede hacer: -Utilizando luz ultravioleta. El método es aplicable para la detección de sustancias capaces de emitir su propia radiación (luminiscencia) en el rango de longitud de onda visible bajo la acción de la radiación UV; mediante reactivos-reveladores. Por ejemplo, la presencia de aminoácidos en la mezcla analizada se puede detectar usando ninhidrina. El cromatograma seco se sumerge en una solución al 0,2% de ninhidrina en acetona y luego se seca. Las manchas correspondientes a los diversos componentes de la mezcla adquieren una apariencia y, por regla general, un color específico para cada sustancia; - utilizando yodo. En este caso, el cromatograma detectado se introduce en un recipiente, en cuyo fondo hay cristales de yodo. Los vapores de yodo se adsorben en las manchas con más fuerza, por lo que se visualizan las manchas. El yodo es un reactivo revelador no específico. Usando reactivos específicos, es posible no solo determinar el número de componentes de una mezcla, sino también identificar las sustancias separadas por el color de las manchas. La cromatografía en papel y en capa fina se lleva a cabo con mayor frecuencia en la llamada variante ascendente descrita anteriormente. Muy a menudo, para mejorar la calidad de los cromatogramas, es necesario utilizar variantes más complejas de cromatografía plana, por ejemplo, de arriba hacia abajo, circular, bidimensional. En la cromatografía descendente en papel o de capa fina, el analito se aplica a la línea de inicio de la placa o tira de papel en la parte superior, y el eluyente se alimenta desde la parte superior en lugar de desde la parte inferior. El efecto positivo, que es mejorar la separación, se debe a la contribución al proceso de separación de la gravedad de los componentes. Tanto la cromatografía upstream como la downstream se pueden realizar en versiones unidimensionales y bidimensionales. A diferencia del proceso de separación unidimensional en lecho plano descrito anteriormente, en la separación cromatográfica bidimensional, la separación de la muestra analizada se realiza primero en un solvente, luego la separación se realiza en la dirección perpendicular a la primera, usando otro solvente, girando el primer cromatograma 90°C. En la cromatografía circular, el analito se aplica como una gota en medio de una placa o una hoja de papel cromatográfico. Aquí también se añaden gota a gota uno o varios disolventes. Esto lleva al hecho de que el cromatograma resultante es un conjunto de puntos radiales. La posición de los puntos (zonas) que forman los componentes separados del analito en un cromatograma plano se caracteriza por los valores de la velocidad relativa de movimiento de los componentes en una capa delgada R fi. Valor experimental R fi definida como la razón de la distancia L I, aprobado I-ésima componente, a la distancia L pasado por el solvente desde la línea de partida hasta la línea frontal (Fig. 1.10): Valor R fi depende de la naturaleza del componente relevante de la muestra analizada, la naturaleza de la fase estacionaria, su espesor, la naturaleza y calidad de la fase móvil, el método de aplicación de la muestra y otros factores, pero siempre R fi
1. Valor R fi es en realidad idéntico al tiempo de retención de una sustancia o su volumen de retención, que caracteriza la velocidad de paso de una sustancia a través de una columna cromatográfica, y puede usarse para la identificación cualitativa de los componentes de la muestra analizada, y el diámetro del punto es idéntico a la altura o área del pico cromatográfico y, por tanto, refleja en cierta medida el contenido cuantitativo de la sustancia. La determinación cuantitativa de la composición de la muestra analizada en el caso más simple puede evaluarse visualmente por la intensidad del color intrínseco de las manchas o la intensidad del brillo fluorescente de las manchas obtenidas durante la detección UV. Para estos fines, la elución de manchas cromatográficas es ampliamente utilizada. En este caso, la mancha obtenida en el cromatograma se recorta o raspa con cuidado, se trata con un disolvente adecuado y la solución resultante se examina mediante el método fisicoquímico adecuado. También puede utilizar el método gravimétrico, en el que se recorta el punto correspondiente del cromatograma y se pesa. La cantidad de sustancia se determina por la diferencia de pesos de papel limpio de la misma superficie y papel con la sustancia. Papel
(BH
) y cromatografía de capa fina
(TLC
) según el mecanismo de separación pertenecen a cromatografía de partición
. En el método HD, el portador es un especial papel cromatográfico
con ciertas propiedades. Fase estacionaria
es agua adsorbida en la superficie y poros del papel (hasta un 20%), móvil - disolvente orgánico, miscible o inmiscible con agua, agua o soluciones electrolíticas. Mecanismo
bastante complicado en el papel. En la fase estacionaria, la sustancia puede retenerse no solo por disolución en el agua adsorbida por el papel, sino también ser adsorbido
celulosa directamente. dibujado en papel componentes compartidos
pasar a la fase móvil y moverse a través de los capilares del papel a diferentes velocidades de acuerdo con coeficiente de distribución interfacial
cada uno de ellos. Al principio cromatografía
Parte de la sustancia del papel pasa a fase móvil
y seguir adelante. Cuando el disolvente orgánico llega a la zona del papel que no contiene el soluto, redistribución
: de la fase orgánica, la sustancia pasa a la fase acuosa, absorbida en papel. Dado que los componentes tienen diferentes afinidad por el sorbente
, al mover el eluyente, se produce la separación: algunas sustancias se retrasan al comienzo del camino, otras se mueven más. Aquí se combinan termodinámica
(establecimiento de una distribución equilibrada de sustancias entre las fases) y cinético
(componentes en movimiento a diferentes velocidades) aspectos de separación. Como resultado, cada componente se concentra en un área específica de la hoja de papel: zonas de componentes individuales
sobre el cromatograma
. El uso de la cromatografía en papel tiene una serie de desventajas significativas: la dependencia del proceso de separación de la composición y propiedades del papel, el cambio en el contenido de agua en los poros del papel con cambios en las condiciones de almacenamiento, una cromatografía muy baja velocidad (hasta varios días), y baja reproducibilidad de los resultados. Estas deficiencias afectan seriamente la difusión de la cromatografía en papel como método cromatográfico. V método CCF
el proceso de separar una mezcla de sustancias se lleva a cabo en una capa delgada sorbente
depositado sobre un sustrato sólido inerte, y proporcionado por el movimiento fase móvil
(solvente) a través del sorbente bajo la acción de fuerzas capilares
.
Pormecanismo de separación
distinguir cromatografía de partición, adsorción e intercambio iónico
. La separación de los componentes se produce en estos casos bien como consecuencia de su diferente coeficiente de distribución entre las dos fases líquidas ( cromatografía de partición
), o debido a la diferente capacidad de adsorción de los compuestos por el adsorbente ( cromatografía de adsorción
). El método de adsorción se basa en diferentes grados de sorción-desorción de los componentes separados en la fase estacionaria. Adsorción
realizado a expensas las fuerzas de van der Waals
, que es la base adsorción física
,
polimolecular
(formación de varias capas de adsorbato en la superficie del adsorbente) y quimisorción
(interacción química de adsorbente y adsorbato). En el caso de usar adsorbentes para TLC como alúmina
o gel de sílice
desempeñar un papel en la separación distribución
, y adsorción
sobre la superficie activa desarrollada del adsorbente (150–750 m2/g). Distribución
componentes de la mezcla se produce entre el agua en la superficie del portador (como adsorbentes
, cómo alúmina
,
almidón
,
celulosa
,
tierra de diatomeas
- y agua
formulario fase estacionaria
), y el solvente moviéndose a través de esta fase estacionaria ( fase móvil
). El componente de la mezcla que es más fácilmente soluble en agua se mueve más lentamente que el que es más fácilmente soluble en la fase móvil. Adsorción
manifiesta en el hecho de que entre transportador
, por ejemplo, óxido de aluminio, y los componentes de la mezcla se fijan equilibrios de adsorción
- para cada componente lo suyo, cuyo resultado es diferente velocidad de viaje
componentes de la mezcla. Se pueden distinguir dos casos extremos: a) la concentración de la sustancia en el adsorbente es cero. La sustancia se disuelve completamente en la fase móvil y es arrastrada por ella (se mueve junto con frente solvente
). b) la sustancia se adsorbe por completo, no interacciona con el disolvente y permanece al principio. En la práctica, con la hábil selección de solvente y adsorbente distribución
compuestos se ubican entre estos casos extremos, y la sustancia gradualmente transferido
de una capa absorbente a otra debido a procesos simultáneos sorción
y desorción
. El solvente que pasa a través del sorbente se llama eluyente
, el proceso de mover una sustancia junto con un eluyente elución
.
A medida que el líquido se mueve sobre la placa, la mezcla de sustancias se separa debido a la acción de fuerzas adsorción
,
distribución
,
intercambio iónico
o una combinación de todos estos factores. Como resultado, separe zonas cromatográficas
componentes de la mezcla, es decir resulta cromatograma
. Selección correcta sorbente
y eluyente
determina la eficiencia de la separación de la mezcla. La movilidad de la sustancia de ensayo depende de su afinidad por el adsorbente y fuerza de elución
(polaridad) del eluyente. A medida que aumenta la polaridad del compuesto, también aumenta su afinidad por el sorbente polar. Al aumentar el grado de adsorción gel de sílice
Los compuestos orgánicos están ordenados en una fila: hidrocarburos.<алкилгалогенидыарены<нитросоединения<простые
эфиры <сложные эфиры<альдегиды<спирты<амины<карбоновые
кислоты. В свою очередь para gel de sílice
los eluyentes se pueden organizar en orden ascendente de "polaridad" ( poder de elución
) y forman una serie de disolventes ( serie eluotrópica
) de acuerdo con datos experimentales: alcanos> benceno> cloroformo> éter dietílico> acetato de etilo> alcoholes С 2 -С 4> agua> acetona> ácido acético> metanol. Por lo tanto, un compuesto polar, el alcohol, se adsorbe con bastante fuerza en el gel de sílice y, por lo tanto, se mueve débilmente bajo la acción de un solvente no polar como el hexano y permanece cerca de la línea de inicio. A su vez, el hidrocarburo aromático no polar bifenilo es notablemente más móvil en hexano, pero incluso aquí, para lograr R
F
aproximadamente 0,5, se necesita un eluyente aprótico más polar, cloruro de metileno. fuerza del eluyente
regular usando mezclas de solventes - vecinos en serie eluotrópica
con diferente polaridad. Actualmente, TLC utiliza principalmente los siguientes adsorbentes
: por separación sustancias lipofílicas
gel de sílice
,
alúmina
,
celulosa acetilada
,
poliamidas
; separar sustancias hidrófilas
celulosa
,
intercambiadores de iones de celulosa
,
tierra de diatomeas
,
poliamidas
. La característica más importante de un adsorbente es su actividad
, es decir. capacidad absorber
(mantener) los componentes de la mezcla a separar. En el extranjero, varias empresas producen gel de sílice
,
tierra de diatomeas
y alúmina
con la adición de un 5% de yeso, que se utiliza para fijar la capa absorbente en la fabricación propia de placas. El sorbente más común es gel de sílice
- ácido silícico hidratado, formado por la acción de ácidos minerales sobre el Na 2 SiO 3 y el secado del sol resultante. Después de moler el sol, se utiliza una fracción de un determinado tamaño de grano (indicado en la placa, normalmente de 5 a 20 micras). gel de sílice
es un sorbente polar
con grupos OH como centros activos. Absorbe fácilmente el agua en la superficie y forma enlaces de hidrógeno. Alúmina
es un adsorbente débilmente básico y se utiliza principalmente para la separación de compuestos débilmente básicos y neutros. La desventaja de las placas sobre óxido de aluminio es la activación obligatoria de la superficie antes de su uso en un armario de secado a alta temperatura (100-150 o C) y la baja capacidad de adsorción de la capa en comparación con el gel de sílice. tierra de diatomeas
- adsorbente obtenido de minerales naturales - tierras de diatomeas. El sorbente tiene propiedades hidrofílicas y una menor capacidad de adsorción de la capa en comparación con el gel de sílice. Celulosa:
Las placas de capa fina recubiertas de celulosa son muy eficaces para separar moléculas orgánicas complejas. El adsorbente consiste principalmente en bolas de celulosa con un diámetro de hasta 50 micras, fijadas en el soporte con almidón. Como en la cromatografía en papel, el ascenso del frente de disolvente es muy lento. Análisis cromatográfico
se lleva a cabo sobre placas industriales de producción checa " Silufol
»
(« Silufol
"") de papel de aluminio, a veces reforzado con cartón, y " Siluplast
» de plástico recubierto con una capa de adsorbentes: gel de sílice LS 5-40 con almidón o yeso como aglutinante (hasta un 10 %), u óxido de aluminio con o sin indicadores fluorescentes. Registros " Silufol
» tienen una alta tasa de elución, sin embargo, se caracterizan por un bajo poder de separación y baja sensibilidad. Durante el almacenamiento, son sensibles a las condiciones (humedad, temperatura, medios agresivos, etc.). Suministro de empresas individuales placas cromatográficas
con una capa de sorbente de espesor diferente (generalmente hasta 0,25 mm), pero estrictamente constante (gel de sílice, celulosa, resina de intercambio iónico), sobre vidrio y sustratos hechos de papel de aluminio, plástico, fibra de vidrio impregnada. Platos " Sorbfil
» (TU 26-11-17-89) se producen en Rusia sobre una base de polímero (tereftalato de polietileno, grado P) o sustrato de aluminio (grado AF) con una capa de trabajo aplicada absorbente de gel de sílice microfraccionado
grados STX-1A y STX-1VE (producidos en la URSS como gel de sílice fraccionado KSKG) con un espesor de 90-120 micrones (hasta 200 micrones), fijados con un aglutinante especial - sílicasol
. Cuando se utiliza sol de ácido silícico (silicazol) como aglutinante, que se transforma en gel de sílice después del calentamiento, las placas de TLC resultantes constan de dos componentes: una capa de gel de sílice y un sustrato. La uniformidad del espesor de la capa absorbente en una placa es de ±5 µm. Ejemplo de designación: "Sorbfil-PTSKh-AF-V-UV (10x10)" - Placas TLC de alto rendimiento sobre un sustrato de aluminio, con fósforo, 10x10 cm. Si se utiliza un sustrato de vidrio (grado C), dichas placas son reutilizables y químicamente resistentes. Su resistencia química está determinada por la resistencia química del gel de sílice. Como resultado, las placas de TLC se pueden tratar repetidamente con reactivos agresivos, por ejemplo, con una mezcla de cromo caliente, lo que elimina las restricciones sobre el uso de reactivos de correlación para la detección puntual y la modificación del sorbente, y permite múltiples (hasta 30 veces o más) regeneración de las placas con una mezcla de cromo. Las placas de vidrio se pueden cortar al tamaño deseado. La resistencia mecánica de la capa adsorbente se puede controlar, proporcionando, por un lado, el transporte y el procesamiento múltiple de las placas y, por otro lado, la posibilidad de extraer capas adsorbentes con sustancias separadas para el posterior lavado de compuestos individuales de la adsorbente y su posterior estudio por métodos instrumentales (espectrometría IR y UV), métodos de difracción de rayos X, RMN, etc.). Las placas difieren en el tamaño de las fracciones (distribución de partículas) del gel de sílice que forma la capa. En placas analíticas (grado A) la fracción es de 5-17 micrones, en altamente eficiente (grado B) - 8-12 micrones. Una distribución más estrecha aumenta la eficiencia de las placas, es decir las manchas de las sustancias a separar se vuelven más compactas (menor tamaño) y por lo tanto se separan mejor cuando el frente del eluyente pasa una distancia más corta. En las obleas rusas, las capas analíticas y de alto rendimiento no difieren mucho, en contraste con las obleas de Merck (Alemania). Se deben utilizar placas de alto rendimiento si las sustancias no se pueden separar en placas analíticas. Las placas de todas las modificaciones se producen con un fósforo (grado UV) con excitación de 254 nm. La vida útil no está limitada, las placas " Sorbfil
» ampliamente probado en el análisis de derivados de aminoácidos, pesticidas, lípidos, antibióticos. El método TLC se lleva a cabo identificación cualitativa
componentes cuantificación
para TLC también es posible, esto requiere aplicar la cantidad exacta de sustancia y adicional estudios densitométricos
con una clara fijación de la intensidad de las manchas. El más común es método semicuantitativo
. Está basado en comparación visual
el tamaño y la intensidad de una mancha componente con las características correspondientes de una serie de manchas de la misma sustancia de diferentes concentraciones ( soluciones estándar de referencia
). Cuando se usa una muestra en la cantidad de 1-5 μg, un método tan simple proporciona una precisión para determinar el contenido de componentes de aproximadamente 5-10%. A menudo, para determinar los componentes de una muestra, es necesario realizar una preparación de la muestra para obtener una mezcla que contenga los compuestos analizados. La preparación de la muestra se basa en la extracción de fármacos de la muestra con disolventes orgánicos ( norte-hexano, éter de petróleo, éter dietílico, cloroformo), purificación del extracto y posterior cromatografía en una fina capa de alúmina o gel de sílice. Hay varias variantes de TLC y BC, que difieren en la forma suministro de solvente
. Dependiendo de la dirección del movimiento de la fase móvil, hay: a)cromatografía ascendente
la fase móvil se vierte en el fondo de la cámara de separación, el papel (placa) se coloca verticalmente; B)cromatografía descendente
la fase móvil se alimenta desde arriba y desciende a lo largo de la capa absorbente de la placa o el papel; v)cromatografía radial
avance horizontal del frente del solvente: la fase móvil es llevada al centro del disco de papel (placa), donde se deposita la mezcla a separar. El más común es elución ascendente
(cromatografía). Parte delantera
eluyente
mientras se mueve de abajo hacia arriba. La elección del solvente (fase móvil) está determinada por la naturaleza del adsorbente y las propiedades de las sustancias a separar. Separación cromatográfica
Los métodos BC y TLC se llevan a cabo en cámara de separación
con tapa atornillada. Una medida cuantitativa de la tasa de transferencia de una sustancia usando un adsorbente y solvente específico es valor R
F
(De inglés. retencion
factor
– coeficiente de retardo, este parámetro es análogo al tiempo de retención). Posición zonas del componente cromatografiado
ajustar al tamaño coeficiente
R
F
igual a la relación entre la velocidad de su zona y la velocidad del frente del solvente. Valor R
F
siempre es menor que la unidad y no depende de la longitud del cromatograma. por la cantidad R
F
influida por diversos factores. Entonces, a bajas temperaturas, las sustancias se mueven más lentamente; la contaminación del solvente, la falta de homogeneidad del adsorbente, los iones extraños en la solución analizada pueden cambiar el valor R
F
hasta 10%. En el sistema seleccionado, los analitos deben tener valores diferentes R
F
y distribuidos a lo largo de toda la longitud del cromatograma. Es deseable que los valores R
F
estaba en el rango de 0.05-0.85. En la práctica, el valor R F
calculada como la razón de la distancia yo
recorrida por la sustancia a la distancia L
pasado por el solvente: R
F
=
l/l
(6.1
)
Por lo general, para el cálculo, elija punto central
(Figura 1). Valor R
F
depende de muchos factores como papel cromatográfico
(su porosidad, densidad, espesor, grado de hidratación) y sorbente
(tamaño de grano, naturaleza de los grupos en la superficie, espesor de la capa, su contenido de humedad, naturaleza de la sustancia, composición de la fase móvil), condiciones experimentales (temperatura, tiempo de cromatografía, etc.). Con la constancia de todos los parámetros cromatográficos, el valor R
F
determinado únicamente por las propiedades individuales de cada componente. Arroz. 1. Determinación de valores en el cromatograma. RF
para componentes A y V, su grado de separación $
y el número de platos teóricos norte
. La eficiencia de BC y TLC también depende de selectividad y sensibilidad
reacciones utilizadas para detectar los componentes de la mezcla analizada. Por lo general, se utilizan reactivos que forman compuestos coloreados con los componentes a determinar: reveladores. Para una más confiable identificación de componentes compartidos
solicitar " testigos
» -soluciones sustancias estándar
(en el mismo solvente que la muestra) que se espera que estén presentes en la muestra. Sustancia estándar
aplicado a la línea de partida junto a la muestra analizada y cromatografiado en las mismas condiciones. En la práctica, a menudo se utiliza un valor relativo: R
F
real
=
R
F
X
/
R
F
estar
(6.2)
donde R
F
estar
también calculado por la fórmula (6.1). Eficiencia
separación cromatográfica
caracterizar número de platos teóricos equivalentes
y ellos altura
. Así, en el método TLC, el número de platos teóricos equivalentes norte
A para componente A la mezcla a separar se calcula mediante la fórmula: norte
A
= 16 (yo
OA
/
a
(A
))
2
(6.3)
Valores yo
OA
y a
(A
)
determinado como se muestra en la Fig. 6.1. Entonces la altura de la placa teórica equivalente H
A
es: H
A
=
yo
OA
/NORTE
=
a
(A
)
2
/ 16
yo
OA
.
(6.4)
La separación es prácticamente posible si R
F
(A)
R
F
(V)
0,1
. Caracterizar la separación de dos componentes. A y V usar grado (criterio) de división Rs
: $
=
l /
(a
(A)
/
2
+
a
(B)
/
2)=
2
l /
(a
(A)
+
a
(B))
(6.5)
donde
yo
distancia entre los puntos centrales de los componentes A y V; a
(A)
y a
(V)
diámetros de punto A y V en el cromatograma (Fig. 6.1). Cuanto más $
, más claramente se separan las manchas de los componentes A y V en el cromatograma. Condiciones cromatografía
se eligen de modo que el valor $
diferente de cero y uno, el valor óptimo $
es 0.3
0.7. Por tasa selectividad de separación
dos componentes A y V usar factor de separación
α
: α
=
yo
B
/
yo
A
(6.6)
Si α = 1, entonces los componentes A y V no están separados.