"High performance liquid chromatography of natural and waste water pollutants"
Introduction
Chapter 1. Basic concepts and classification of liquid chromatography methods
1.1 Apparatus for liquid chromatography
Chapter 2. The essence of HPLC
2.1 Application
Chapter 3. Examples of using HPLC in the analysis of environmental objects
Chapter 4 HPLC Instrumentation
Literature
Appendix
Introduction
Chromatographic Methods are often indispensable for the identification and quantification of organic compounds with a similar structure. The most widely used for routine analysis of environmental pollutants are gas and high performance liquid chromatography. Gas chromatographic analysis of organic pollutants in drinking and waste waters was initially based on the use of packed columns, later quartz capillary columns also became widespread. The internal diameter of capillary columns is usually 0.20-0.75 mm, length - 30-105 m. Optimum results in the analysis of contaminants in water are most often achieved when using capillary columns with different film thicknesses made of methylphenyl silicones with a content of phenyl groups of 5 and 50% . The sample injection system often becomes a vulnerable point in chromatographic techniques using capillary columns. Sample injection systems can be divided into two groups: universal and selective. The universal ones include injection systems with and without splitting the flow, “cold” injection into the column and evaporation with temperature programming. Selective injection uses purge with intermediate trapping, headspace analysis, etc. When using universal injection systems, the entire sample enters the column, with selective injection, only a certain fraction is introduced. The results obtained with selective injection are significantly more accurate, since the fraction that entered the column contains only volatile substances, and the technique can be fully automated.
Gas chromatographic detectors used in pollutant monitoring are often divided into universal detectors, which respond to each component in the mobile phase, and selective detectors, which react to the presence in the mobile phase of a certain group of substances with similar chemical characteristics. The universal ones include flame ionization, atomic emission, mass spectrometric detectors and infrared spectrometry. Selective detectors used in water analysis are electron-capture (selective to substances containing halogen atoms), thermionic (selective to nitrogen- and phosphorus-containing compounds), photoionization (selective to aromatic hydrocarbons), electrolytic conductivity detector (selective to compounds, containing halogen, sulfur and nitrogen atoms). The minimum detectable amounts of substances range from nanograms to picograms per second.
High Performance Liquid Chromatography(HPLC) is an ideal method for the determination of a large number of thermally unstable compounds that cannot be analyzed using gas chromatography. Currently, modern agrochemicals, including methyl carbonates and organophosphorus insecticides, and other non-volatile substances, often become objects of analysis by liquid chromatography. High performance liquid chromatography (HPLC) is gaining popularity among other methods used in environmental monitoring, also because it has bright prospects in terms of automating sample preparation.
CHAPTER 1. BASIC CONCEPTS AND CLASSIFICATION OF LIQUID CHROMATOGRAPHY METHODS
Liquid chromatography is divided into several classes depending on the type of stationary phase support. The simple instrumentation of paper and thin layer chromatography led to the widespread use of these methods in analytical practice. However, the great possibilities of column liquid chromatography stimulated the improvement of equipment for this classical method and led to the rapid introduction of HPLC. Passing the eluent through the column under high pressure made it possible to sharply increase the analysis rate and significantly increase the separation efficiency due to the use of a finely dispersed sorbent. The HPLC method currently makes it possible to isolate, quantitatively and qualitatively analyze complex mixtures of organic compounds.
According to the mechanism of interaction of the separated substance (eluate) with the stationary phase, adsorption, distribution, ion-exchange, size-exclusion, ion-pair, ligand-exchange and affinity chromatography are distinguished.
Adsorption chromatography. Separation by adsorption chromatography is carried out as a result of the interaction of the substance to be separated with an adsorbent, such as aluminum oxide or silica gel, which have active polar centers on the surface. The solvent (eluent) is a non-polar liquid. The mechanism of sorption consists in a specific interaction between the polar surface of the sorbent and the polar (or capable of being polarized) regions of the molecules of the analyzed component (Fig. 1).
Rice. 1. Adsorption liquid chromatography.
Partition chromatography. In the distributive version of liquid chromatography, the separation of a mixture of substances is carried out due to the difference in their distribution coefficients between two immiscible phases - the eluent (mobile phase) and the phase located on the sorbent (stationary phase).
At normal-phase Partition liquid chromatography uses a non-polar eluent and polar groups grafted onto the surface of a sorbent (most often silica gel). Substituted alkylchlorosilanes containing polar groups, such as nitrile, amino group, etc., are used as silica gel surface modifiers (bonded phases) (Fig. 2). The use of bonded phases makes it possible to finely control the sorption properties of the surface of the stationary phase and achieve high separation efficiency.

Rice. 2. Partition chromatography with bonded phase (normal phase variant).
reversed phase liquid chromatography is based on the distribution of mixture components between the polar eluent and nonpolar groups (long alkyl chains) grafted onto the sorbent surface (Fig. 3).

Rice. 3. Partition chromatography with bonded phase (reversed-phase version).
Less widely used is a variant of supported phase liquid chromatography, in which a liquid stationary phase is applied to a stationary support.
Exclusive (gel penetrating) Chromatography is a variant of liquid chromatography in which the separation of substances occurs due to the distribution of molecules between the solvent located in the pores of the sorbent and the solvent flowing between its particles.
affine Chromatography is based on specific interactions of separated proteins (antibodies) with substances (antigens) grafted onto the surface of a sorbent (synthetic resin) that selectively form complexes (conjugates) with proteins.
Ion-exchange, ion-pair, ligand-exchange chromatography are used mainly in inorganic analysis.
Basic parameters of chromatographic separation.
The main parameters of chromatographic separation are the retention volume and the retention time of the mixture component (Fig. 4).
The retention time tR is the time elapsed from the moment the sample is injected into the column until the maximum of the corresponding peak is reached. Multiplying the retention time by the eluent volume velocity F, we get the retention volume VR:
The corrected retention time is the time elapsed from the moment the peak of the non-sorbable component appears to the peak of the corresponding compound:
tR" = tR - t0 ;
The normalized or corrected retention volume is the retention volume corrected for column dead volume V0, i.e. the retention volume of the non-sorbable component:
VR" = VR - V0;
The retention characteristic is also the capacitance factor k", defined as the ratio of the mass of the substance in the stationary phase to the mass of the substance in the mobile phase: k" = mn / mp;
The value of k" is easy to determine from the chromatogram:

The most important parameters of chromatographic separation are its efficiency and selectivity.
The efficiency of the column, measured by the height of the theoretical plates (HETP) and inversely proportional to their number (N), is higher, the narrower the peak of the substance emerging at the same retention time. The efficiency value can be calculated from the chromatogram using the following formula:
N = 5.54. (tR / 1/2) 2 ,
where tR- holding time,
w 1/2 - peak width at half height
Knowing the number of theoretical plates per column, the column length L, and the average sorbent grain diameter dc, it is easy to obtain the values of the theoretical plate equivalent height (HETP) and the reduced height (PETP):
HETP = L/N PHETP = HETP/d c
These characteristics make it possible to compare the efficiency of columns of different types, to evaluate the quality of the sorbent and the quality of filling the columns.
The selectivity of the separation of two substances is determined by the equation:
When considering the separation of a mixture of two components, the degree of separation RS is also an important parameter:
 ;
;
Peaks are considered resolved if the RS value is greater than or equal to 1.5.
The main chromatographic parameters relate the following equation for resolution:
 ;
;
The factors that determine separation selectivity are:
1) the chemical nature of the sorbent;
2) the composition of the solvent and its modifiers;
3) chemical structure and properties of the components of the mixture to be separated;
4) column temperature
1.1 Apparatus for liquid chromatography
In modern liquid chromatography, instruments of varying degrees of complexity are used - from the simplest systems to high-end chromatographs equipped with various additional devices.
On fig. Figure 4 shows a block diagram of a liquid chromatograph containing the minimum required set of components, in one form or another, present in any chromatographic system.

Rice. 4. Block diagram of a liquid chromatograph.
The pump (2) is designed to create a constant solvent flow. Its design is determined primarily by the operating pressure in the system. For operation in the range of 10-500 MPa, plunger (syringe) or piston type pumps are used. The disadvantage of the first is the need for periodic stops for filling with eluent, and the second is the great complexity of the design and, as a result, the high price. For simple systems with low operating pressures of 1-5 MPa, inexpensive peristaltic pumps are successfully used, but since it is difficult to achieve a constant pressure and flow rate, their use is limited to preparative tasks.
The injector (3) ensures that a sample of the mixture of separated components is injected into the column with a sufficiently high reproducibility. Simple "stop-flow" sample injection systems require the pump to be stopped and are therefore less convenient than Reodyne's loop pipettes.
The HPLC columns (4) are thick-walled stainless steel tubes capable of withstanding high pressure. An important role is played by the density and uniformity of packing the column with a sorbent. For low pressure liquid chromatography, thick-walled glass columns are successfully used. Temperature constancy is ensured by thermostat (5).
Detectors (6) for liquid chromatography have a flow cell in which some property of the flowing eluent is continuously measured. The most popular types of general purpose detectors are refractometers, which measure the refractive index, and spectrophotometric detectors, which measure the absorbance of a solvent at a fixed wavelength (usually in the ultraviolet region). The advantages of refractometers (and the disadvantages of spectrophotometers) include low sensitivity to the type of compound being determined, which may not contain chromophore groups. On the other hand, the use of refractometers is limited to isocratic systems (with a constant eluent composition), so the use of a solvent gradient is not possible in this case.
HPLC columns, which are most commonly used in environmental pollutant analysis, are 25 cm long and 4.6 mm inside diameter, and are filled with 5-10 µm spherical silica gel particles grafted with octadecyl groups. In recent years, columns with smaller internal diameters filled with smaller particles have appeared. The use of such columns reduces the consumption of solvents and the duration of the analysis, increases the sensitivity and separation efficiency, and also facilitates the problem of connecting columns to spectral detectors. Columns with an internal diameter of 3.1 mm are equipped with a safety cartridge (precolumn) to increase the service life and improve the reproducibility of the analyses.
As detectors in modern HPLC instruments, a UV detector on a diode array, fluorescence, and electrochemical detectors are usually used.
It should be borne in mind that in practical work, separation often proceeds not by one, but by several mechanisms simultaneously. So, exclusion separation can be complicated by adsorption effects, adsorption - distribution, and vice versa. In this case, the greater the difference in the substances in the sample in terms of the degree of ionization, basicity or acidity, in terms of molecular weight, polarizability, and other parameters, the greater the likelihood of a different separation mechanism for such substances.
In practice, "reversed-phase" (partition) chromatography, in which the stationary phase is not polar, but the mobile phase is polar (i.e., the reverse of "straight-phase" chromatography), has become most widespread.
In most laboratories around the world, a group of 16 priority PAHs are analyzed by HPLC or CMS.
CHAPTER 2. ESSENCE OF HPLC
In high performance liquid chromatography (HPLC), the nature of the processes occurring in the chromatographic column is generally identical to the processes in gas chromatography. The difference is only in the use of a liquid as a stationary phase. Due to the high density of liquid mobile phases and the high resistance of the columns, gas and liquid chromatography differ greatly in instrumentation.
In HPLC, pure solvents or their mixtures are usually used as mobile phases.
To create a stream of pure solvent (or mixtures of solvents), called eluent in liquid chromatography, pumps are used, which are part of the chromatograph hydraulic system.
Adsorption chromatography is carried out as a result of the interaction of a substance with adsorbents, such as silica gel or aluminum oxide, which have active centers on the surface. The difference in the ability to interact with the adsorption centers of different sample molecules leads to their separation into zones in the process of moving with the mobile phase through the column. The division of the component zones achieved in this case depends on the interaction with both the solvent and the adsorbent.
Silica gel adsorbents with different volumes, surfaces, and pore diameters find the greatest application in HPLC. Aluminum oxide and other adsorbents are used much less frequently. The main reason for this:
Insufficient mechanical strength, which does not allow packaging and use at elevated pressures typical for HPLC;
silica gel compared to aluminum oxide has a wider range of porosity, surface and pore diameter; a significantly higher catalytic activity of aluminum oxide leads to a distortion of the analysis results due to the decomposition of the sample components or their irreversible chemisorption.
HPLC detectors
High performance liquid chromatography (HPLC) is used to detect polar non-volatile substances, which for some reason cannot be converted into a form convenient for gas chromatography, even in the form of derivatives. Such substances, in particular, include sulfonic acids, water-soluble dyes and some pesticides, such as phenyl-urea derivatives.
Detectors:
UV - diode array detector. The "matrix" of photodiodes (there are more than two hundred of them) constantly registers signals in the UV and visible region of the spectrum, thus ensuring the recording of UV-B spectra in the scanning mode. This makes it possible to continuously record, at high sensitivity, undistorted spectra of components rapidly passing through a special cell.
Compared to single-wavelength detection, which does not provide information about the "purity" of the peak, the ability to compare the full spectra of the diode array provides an identification result with a much greater degree of certainty.
Fluorescent detector. The great popularity of fluorescent detectors is due to the very high selectivity and sensitivity, and the fact that many environmental pollutants fluoresce (for example, polyaromatic hydrocarbons).
An electrochemical detector is used to detect substances that are easily oxidized or reduced: phenols, mercaptans, amines, aromatic nitro and halogen derivatives, aldehydes, ketones, benzidines.
Chromatographic separation of the mixture on the column due to the slow advance of the PF takes a long time. To speed up the process, chromatography is carried out under pressure. This method is called high performance liquid chromatography (HPLC).
Modernization of the equipment used in classical liquid column chromatography has made it one of the promising and modern methods of analysis. High performance liquid chromatography is a convenient method for separating, preparatively isolating, and performing qualitative and quantitative analysis of non-volatile thermolabile compounds of both low and high molecular weight.
Depending on the type of sorbent used in this method, 2 variants of chromatography are used: on a polar sorbent using a non-polar eluent (direct phase option) and on a non-polar sorbent using a polar eluent - the so-called reverse-phase high-performance liquid chromatography (RPHLC).
When the eluent passes to the eluent, the equilibrium under RPHLC conditions is established many times faster than under the conditions of polar sorbents and non-aqueous PFs. As a result of this, as well as the convenience of working with water and water-alcohol eluents, RPHLC has now gained great popularity. Most HPLC analyzes are carried out using this method.
Detectors. Registration of the output from the column of a separate component is performed using a detector. For registration, you can use the change in any analytical signal coming from the mobile phase and related to the nature and amount of the mixture component. Liquid chromatography uses such analytical signals as light absorption or light emission of the exiting solution (photometric and fluorimetric detectors), refractive index (refractometric detectors), potential and electrical conductivity (electrochemical detectors), etc.
The continuously detected signal is recorded by the recorder. The chromatogram is a sequence of detector signals recorded on the recorder tape, generated when individual components of the mixture exit the column. In the case of separation of the mixture, individual peaks are visible on the external chromatogram. The position of the peak on the chromatogram is used for the purpose of identification of the substance, the height or area of the peak - for the purpose of quantitative determination.
2.1 Application
HPLC finds the widest application in the following areas of chemical analysis (objects of analysis where HPLC has practically no competition are highlighted):
· Food quality control - tonic and flavor additives, aldehydes, ketones, vitamins, sugars, dyes, preservatives, hormones, antibiotics, triazine, carbamate and other pesticides, mycotoxins, nitrosoamines, polycyclic aromatic hydrocarbons, etc.
· Environmental protection - phenols, organic nitro compounds, mono- and polycyclic aromatic hydrocarbons, a number of pesticides, major anions and cations.
· Criminalistics - drugs, organic explosives and dyes, potent pharmaceuticals.
· Pharmaceutical industry - steroid hormones, practically all products of organic synthesis, antibiotics, polymer preparations, vitamins, protein preparations.
Medicine - the listed biochemical and medicinal substances and their metabolites in biological fluids (amino acids, purines and pyrimidines, steroid hormones, lipids) in the diagnosis of diseases, determining the rate of excretion of drugs from the body for the purpose of their individual dosage.
· Agriculture - determination of nitrate and phosphate in soils to determine the required amount of fertilizers, determination of the nutritional value of feed (amino acids and vitamins), analysis of pesticides in soil, water and agricultural products.
Biochemistry, bioorganic chemistry, genetic engineering, biotechnology - sugars, lipids, steroids, proteins, amino acids, nucleosides and their derivatives, vitamins, peptides, oligonucleotides, porphyrins, etc.
· Organic chemistry - all stable products of organic synthesis, dyes, thermolabile compounds, non-volatile compounds; inorganic chemistry (practically all soluble compounds in the form of ions and complex compounds).
· Quality control and safety of food products, alcoholic and non-alcoholic beverages, drinking water, household chemicals, perfumes at all stages of their production;
determination of the nature of pollution at the site of a man-made disaster or emergency;
detection and analysis of narcotic, potent, poisonous and explosive substances;
determination of the presence of harmful substances (polycyclic and other aromatic hydrocarbons, phenols, pesticides, organic dyes, ions of heavy, alkaline and alkaline earth metals) in liquid effluents, air emissions and solid waste from enterprises and in living organisms;
· monitoring of processes of organic synthesis, oil and coal processing, biochemical and microbiological productions;
analysis of soil quality for fertilization, the presence of pesticides and herbicides in soil, water and products, as well as the nutritional value of feed; complex research analytical tasks; obtaining a micro amount of ultrapure substance.
CHAPTER 3. EXAMPLES OF USE OF HPLC IN THE ANALYSIS OF ENVIRONMENTAL OBJECTS
HPLC - a method for monitoring PAHs in environmental objects
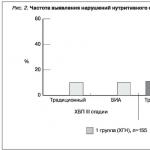
For polycyclic aromatic hydrocarbons (PAHs), ecotoxicants of the 1st hazard class, extremely low levels of maximum permissible concentrations (MACs) in natural objects have been established. The determination of PAHs at the MPC level and below is one of the very complex analytical tasks and high-tech analysis methods (GC-MS, GC, HPLC) are used to solve them. When choosing a method for monitoring, in addition to the main characteristics under consideration - sensitivity and selectivity, expressness and economy are added, because. monitoring involves serial analysis. The HPLC variant on short, small diameter columns meets these requirements to a large extent. Using this method, the authors developed and certified methods for monitoring benzo[a]pyrene in three natural media: aerosol, snow cover, and surface waters. The methods are characterized by: simple unified sample preparation, including extraction of PAHs with organic solvents and concentration of the extract, direct introduction of a concentrated extract into a chromatographic column, the use of multiwavelength photometric detection in the UV region of the spectrum, identification of PAH peaks in chromatograms using two parameters, retention time and spectral ratio . The total error does not exceed 10% when determining benz[a]pyrene in aerosol in the concentration range from 0.3 to 450 ng/m up to 50 μg / m 2. For the case of simultaneous determination of priority PAHs (up to 12 compounds) and registration of inhomogeneous peaks of analytes, it was proposed to reseparate the extract with a change in the selectivity of the mobile phase, the detection wavelength, and the column temperature, taking into account the individual properties of the PAH being determined.
1 . Ambient air quality. Mass concentration of benzo[a]pyrene. The procedure for performing measurements by the HPLC method. Certificate of attestation MVI No. 01-2000.
2 . The quality of surface and treated wastewater. Mass concentration of benzo[a]pyrene. The procedure for performing measurements by the HPLC method. Certificate of attestation MVI No. 01-2001.
3 . Snow cover quality. Mass concentration of benzo[a]pyrene. The procedure for performing measurements by the HPLC method. Certificate of attestation MVI No. 02-2001.
Removal of Aniline from Aqueous Solutions Using Wastes of Aluminothermic Recovery of Rolled Copper Scale
The problem of removing hydrocarbons from wastewater is an urgent task. In many chemical, petrochemical and other industries, aniline and its derivatives are formed, which are toxic substances. Aniline is a highly toxic substance, MPC - 0.1 mg / m 3. Aniline and its derivatives are soluble in water and therefore cannot be removed by gravitational settling.
One of the best methods of wastewater treatment from organic pollutants is the use of inorganic and organic adsorbents capable of regeneration (aluminosilicates, modified clays, wood, fibers, etc.) and incapable of regeneration (activated carbon, macroporous polymeric materials, etc.). ).
Regenerated adsorbents can remove organic substances of different polarity from water. The search for effective adsorbents is an urgent task.
This report presents the results of a study in the field of using the milled copper scale of the Yerevan Cable Plant (OPMOERKZ) as aniline sorbents.
Chromatographic studies were carried out on an HPLC chromatograph / high performance liquid chromatography / systems (Waters 486 - detector, Waters 600S - controller, Waters 626 - Pump), on a 250 x 4 mm column filled with sorbents under study, mobile phase rate 1 ml/m / mobile phase are the solvents we are studying/, the detector is UV-254. UV spectroscopic analysis was carried out on a Specord-50 spectrophotometer, the spectra were obtained using the ASPECT PLUS computer program.
Precisely weighed portions of sorbents were added to certain volumes of aniline in water, the initial concentrations of which varied. The mixture was thoroughly shaken for 6 h. Then the sample was left to settle. Adsorption is completed in almost 48 hours. The amount of precipitated aniline was determined by UV spectrophotometric as well as refractive analysis.
At first, the adsorption properties of OPMOEPKZ were studied during the removal of aniline from a solution in carbon tetrachloride. It turned out that aniline absorbs sorbent 3 best of all (table).
Measurements were also carried out for aqueous solutions of aniline at concentrations of 0.01-0.0001 mol/l. The table shows data on a 0.01 M solution.
Absorption of aniline by various sorbents from 0.01 M aqueous solution of aniline at 20°C
Previously, it was found that adsorption within the indicated concentration ranges increases and depends linearly on the refractive index. The amount of aniline was determined from the refractive index versus molar concentration plot and corrected for both liquid chromatography and UV spectral analysis.
Sorbent 3 is the most active for aqueous solutions. The amount of adsorbed pollutant was calculated as the difference between the total amount of pollutant added to the initial solution and its residue in the final solution.
Methods for determining PAHs in environmental objects
Typically, gas chromatography (GC) and high performance liquid chromatography (HPLC) methods are used to determine PAHs. separation of the main 16 PAHs, sufficient for quantitative analysis, is achieved using either capillary columns in gas chromatography or high-performance columns used in HPLC. It must be remembered that a column that separates well the calibration mixtures of sixteen PAHs does not guarantee that they will also be well separated against the background of accompanying organic compounds in the samples under study.
In order to simplify the analysis, as well as to achieve high quality of the results obtained, most analytical procedures contain the stage of preliminary isolation (separation) of PAHs from other groups of related compounds in samples. The most common methods used for this purpose are low-pressure liquid chromatography in liquid-solid or liquid-liquid systems using adsorption mechanisms, such as using silica gel or alumina, sometimes mixed mechanisms are used, such as adsorption and exclusion using Sephadex.
The use of pretreatment of samples makes it possible to avoid the influence of:
Completely non-polar compounds such as aliphatic hydrocarbons;
Moderately and strongly polar compounds, for example, phthalane, phenols, polyhydric alcohols, acids;
High molecular weight compounds such as, for example, resins.
Two types of detectors are mainly used in high performance liquid chromatography (HPLC): a fluorimetric detector or a photodiode bar spectrophotometric detector. The limit of detection of PAHs in fluorimetric detection is very low, which makes this method particularly suitable for the determination of trace amounts of polyaromatic compounds. However, classical fluorimetric detectors provide virtually no information about the structure of the compound under study. Modern designs make it possible to record fluorescence spectra that are characteristic of individual compounds, but they have not yet become widespread in the practice of routine measurements. A spectrophotometric detector with a photodiode line (PDL) makes it possible to record absorption spectra in the UV and visible spectral range, these spectra can be used for identification. Similar information can be obtained using fast scanning detectors.
When choosing an analytical technique for the separation, identification and quantification of these PAHs, the following conditions must be taken into account:
The level of determined contents in the studied samples;
The number of related substances;
Applied analytical procedure (measurement technique);
Possibilities of the serial equipment.
Development of a Method for Determination of Alkaline Earth Elements and Magnesium by Ion High Performance Liquid Chromatography
The development and improvement of methods that allow solving problems of water analysis is an important problem in analytical chemistry. The development of high-performance high-pressure liquid chromatography stimulated the development of a new direction in ion-exchange chromatography, the so-called ion chromatography. The synthesis of sorbents for ion chromatography is difficult, since quite a lot of requirements are imposed on them. Due to the lack of commercially available high-performance cation exchangers, a dynamically modified reverse phase was used, for which a modifier was synthesized: N-hexadecyl-N-decanoyl-para-amino-benoylsulfonic acid ethyl-diisopropylammonium (DGDASC), where the hydrophobic amine containing the SO 3 - group, capable of cation exchange. After passing the modifier solution, the absorption at l = 260 nm reached 6.4 units of optical density (° E) reaching a plateau. The calculated ion exchange capacity is 15.65 µmol. Since the cations of alkaline earth elements and magnesium do not absorb in the UV region of the spectrum, indirect UV detection was used using the synthesized UV absorbing eluent 1,4-dipyridinium butane bromide (DPB bromide). Since halogen ions destroy the steel parts of the column, the bromide ion of 1,4-dipyridinium butane was replaced by an acetate ion. When the column is washed with eluent, the counterion of the modifier, ethyldiisopropylammonium, is replaced by the UV-absorbing ion 1,4-dipyridiniumbutane. Separation of cations was carried out at the optimal wavelength l = 260 nm on a scale of 0.4 A in the “scale folding” mode; the polarity of the recorder was reversed. The separation of all the studied cations was achieved with the introduction of a complexing additive - oxalic acid. The detection limits of Mg 2+ , Ca 2+ , Sr 2+ , Ba 2+ are 8 μg/l; 16 µg/l; 34 µg/l; 72 µg/l, respectively. Under the selected conditions, tap water was analyzed, the content of Ca 2+ in which is 10.6 +1.9 mg-ion/l, Mg 2+ -2.5 + mg-ion/l. The reproducibility error does not exceed -2.2% for Ca 2+ and 1.4% for Mg 2+.
Analysis of cadmium complexes in the environment
To study the mechanisms of migration of heavy metals in the biosphere, data on the chemical forms of the existence of metals in nature are needed. Difficulties in the analysis of compounds of one of the most toxic metals - cadmium - are associated with the fact that it forms unstable complexes, and when trying to isolate them, natural equilibria are distorted. In this work, cadmium compounds in soil and plants were studied using a technique based on the chromatographic separation of extracts followed by the identification of components by chemical analysis. This approach made it possible not only to identify the chemical forms of cadmium, but also to trace their transformations in environmental objects.
OH-groups of carbohydrates and polyphenols (including flavonoids), C=O, phosphates, NH 2 , NO 2 , SH-groups are coordinated with cadmium in biosphere objects. For the purposes of this study, a set of model ligands representing these classes of compounds was compiled. The interaction of model ligands with water-soluble cadmium salts was studied by UV spectroscopy and HPLC.
To isolate cadmium compounds, extraction with specially selected (not forming complexes with Cd) solvents was used. In this way, cadmium can be separated from all heavy metals, except for its close chemical analogue, zinc. Cadmium- and zinc-containing peaks in the chromatograms of the obtained extracts were detected by binding metals in the form of their dithizonates. To separate from zinc, the difference in the stability of the Cd and Zn complexes at pH 6–8 was used. The isolated Cd compounds were identified by HPLC with pH changes during elution. The analysis of cadmium compounds with the components of soils and plant tissues was performed, and the substances produced by plants in response to an increase in the intake of cadmium from the soil were identified. It has been shown that flavonoids, in particular tricine, are protective agents in cereals, alkoxy derivatives of cysteine in legumes, and both polyphenols and thiols in cruciferous plants.
CHAPTER 4. HPLC EQUIPMENT
SERIES ACCELA
The new ACCELA Ultra High Performance Liquid Chromatograph is capable of operating over a wide range of flow rates and pressures, providing both typical HPLC separations on conventional columns and ultra-fast and efficient separations on columns with a sorbent particle size of less than 2 µm at ultra-high pressures (greater than 1000 atm.).
The system includes a quaternary gradient inert pump capable of delivering pressures in excess of 1000 atm and with a delay volume of only 65 µl, providing high speed chromatographic separations. Autosampler ACCELA capable of operating in a sample injection cycle of 30 seconds and provides the highest injection reproducibility. Diode Array Detector Accela PDA with a minimized flow cell volume (2 µl) is optimized for high-speed chromatography, uses patented LightPipe technology and maintains the symmetrical peak shape that comes with a flawless chromatography system and columns.
The system pairs perfectly with mass spectrometers to create the most powerful and best LC/MS systems available in the world.
1.9 µm UHP columns available from Thermo Electron for all applications
SERIES TSP
The modular principle of construction of HPLC instruments allows the customer to flexibly complete equipment for solving any analytical problems, and when they change, it can be quickly and economically modified. The wide range of modules includes pumps from isocratic to quaternary gradient, from micro-column to semi-preparative, all available detectors, sample injection systems from manual injectors to autosamplers with the ability to handle any sample, powerful software for processing measurement results and managing all modules of the system. All modules are certified according to CSA, TUF/GS, FCC(EMI), VDE (EMI), ISO-9000, they are compact, have a modern design, are easy to operate, equipped with a built-in display and self-diagnostic system, allow you to create and store task methods in memory parameters. They meet the criteria of "Exemplary Laboratory Practice" (GLP) and are listed in the Register of Measuring Instruments of the Russian Federation. Measurement protocols are issued in accordance with the Pharmacopoeias of England, USA, Germany and France.
TSP modular systems are characterized by the highest reliability and stability in operation.
The combination of modules provides the analyst with all the advantages of an integrated system on the one hand and the flexibility of a modular system on the other. Whichever field of application of High Performance Liquid Chromatography (HPLC) - pharmacology, biotechnology, environmental analysis, clinical analysis, food and beverage analysis, petrochemical and chemical analysis - this instrument is used, it is always optimally configured in order to meet the highest requirements.
Both research and high performance routine systems provide:
Highly efficient solvent degassing
Ability to work with small and ultra-small sample quantities
Highest sensitivity, both with UV/VIS detector and diode array (with the famous LightPipe technology with a choice of 1 or 5 cm optical path length)
Working with different columns
Highest Quantitative Accuracy
Possibility of automatic work with different sample volumes
RMS retention time error less than 0.3%
The minimum working area occupied by the system
Highest reliability and parameter stability.
Surveyor LC Pump- An HPLC pump with the best retention time reproducibility of any 4-component gradient pump available in the world. An integrated quad-channel vacuum degasser and pulsation damper provide excellent baseline stability for maximum quantitation sensitivity and accuracy.
The autosampler provides the highest performance and analysis flexibility. A wide range of sample trays, from standard vials to 96- and 384-well microplates, covers the needs of virtually all applications. The new technology provides virtually no loss of sample injection, virtually 5 µl of sample is injected with an autosampler from a total sample volume of 5 µl.
SURVEYOR
UV/Visual Detector and PDA (Diode Array Detector)
Surveyor UV/Vis- variable wavelength ultraviolet and visible light detector is a combination of economy and reliability with the highest sensitivity of LightPipe technology. A wide selection of flow cells makes this detector versatile for all applications from those using capillary or microcolumn chromatography to semi-preparative and preparative.
Surveyor PDA The detector is the most sensitive among all HPLC diode array detectors. Dual-lamp source optics seamlessly cover the entire wavelength range from 190 to 800 nm. The fiber optic beamformer provides excellent optical resolution without sacrificing sensitivity.
Surveyor RI refractometric detector with a minimum volume thermostated cuvette with full electronic control from a computer.
Surveyor FL fluorometric scanning detector with the highest sensitivity and detection capability for fluorescence, chemiluminescence and phosphorescence.
A wide range of autosamplers allows you to work with both conventional vials and 96-position plates, widely used in biochemistry and clinical practice. Handling is facilitated by the use of similar SPE sample preparation plates.
400 Electric drive, Valco loop (20 µl - standard) with the possibility of partial filling.
Carousel 96 samples.
Electric drive, column thermostat, Valco loop (100 µl - standard) with the possibility of partial filling. AutoMix mode for sample preparation. Sample carousel: 84 x 2 ml (samples) + 3 x 10 ml (reagents). Built-in column thermostat. 420
Loop autosampler for research work with the ability to work in the modes of full, partial filling and introduction of microliter samples. A wide range of carousels (standard - 96 samples).
Tablet autosampler for 96- and 384-position plates. Injection of the sample into the pressure loop, the possibility of introducing samples of less than 1 µl. Possibility to install a tablet feeder. HPLC
Major manufacturers of HPLC equipment
· Waters - high performance chromatography, mass spectrometry, columns, solid phase extraction;
Varian, Inc. - chromatographs and columns, accessories for solid phase extraction;
· Agilent Technologies - chromatographs and columns;
· Hypersil - columns and sorbents.
· Merck KGaA - TLC plates and accessories for TLC, columns, sorbents mobile phases for HPLC, accessories for solid phase extraction
· Dionex - equipment and columns for HPLC, especially for ion chromatography.
Literature
1.Pilipenko A.T., Pyatnitsky I.V. Analytical chemistry. In two books: kn..1 - M .: Chemistry, 1990, -480s.
1. Pilipenko A.T., Pyatnitsky I.V. Analytical chemistry. In two books: kn..2 - M .: Chemistry, 1990, -480s.
2. Vasiliev V.P. Analytical chemistry. At 2 pm Part 2. Physical and chemical methods of analysis: Proc. for Khimko - technol. specialist. universities. - M .: Higher. school, 1989. - 384p.
3. Hydrochemical materials. Volume 100. Methods and technical means of operational monitoring of surface water quality. L.: Gidrometeo-izdat, 1991. - 200p.
4. Lurie Yu.Yu. Analytical chemistry of industrial wastewater / Yu.Yu. Lurie; M.: Chemistry Yu, 1984. - 448s.
5. Ewing G. Instrumental methods of chemical analysis / Per. from English. M.: Mir, 1989. - 348 p.
6. Gorelik D.O., Konopelko L.A., Pankov E.D. Environmental monitoring. In 2 vols. St. Petersburg: Christmas. 2000. - 260 p.
7. Aivazov B.V. Introduction to chromatography. M.: Higher. school, 1983. - 450 p.
8. Goldberg K.A., Vigdergauz M.S. Introduction to gas chromatography. M.: Chemistry, 1990. - 329 p.
9. Stolyarov B.V. and others // Practical gas and liquid chromatography. St. Petersburg: St. Petersburg State University, 1998. - S. 81.
11. Gorshkov A.G., Marinaite I.I. HPLC - a method for monitoring PAHs in environmental objects
12. Torosyan G. O., Martirosyan V. A., Aleksanyan A. R., Zakaryan M. O. Removal of aniline from aqueous solutions using waste products of aluminothermal reduction of rolling copper scale
13. L.A. Turkina, G.N. Koroleva Development of a method for the determination of alkaline earth elements and magnesium by ion high performance liquid chromatography
14. Dultseva G.G., Dubtsova Yu.Yu., Skubnevskaya G.I. Analysis of cadmium complexes in the environment
Appendix
DETERMINATION OF CLOMAZONE IN WATER BY CHROMATOGRAPHIC METHODS
METHODOLOGICAL INSTRUCTIONS MUK 4.1.1415-03
1. Prepared by: Federal Scientific Center for Hygiene. F.F.
Erisman; Moscow Agricultural Academy. K.A.
Timiryazev; with the participation of the Department of State Sanitary and Epidemiological Surveillance of the Ministry of Health of Russia. The developers of the methodology are listed at the end.
3. Approved by the Chief State Sanitary Doctor
Russian Federation, First Deputy Minister of Health of the Russian Federation, acad. RAMS G.G. Onishchenko June 24, 2003
5. Introduced for the first time.
1. Introduction
Manufacturer: FMS (USA).
Trade name: COMMAND.
Active ingredient: clomazone.
2-(2-chlorobenzyl)-4,4-dimethyl-3-isoxalidin-3-one(IUPAC)
Light brown viscous liquid.
Melting point: 25 -C.
Boiling point: 275 -C.
Vapor pressure at 25 -C: 19.2 MPa.
Partition coefficient n-octanol/water: K logP = 2.5.
Highly soluble in acetone, hexane, ethanol, methanol,
chloroform, dichloromethane and acetonitrile; solubility in water -
1.10 g/cu. dm. Stable at room temperature for at least 2 years, at 50 -C - at least 3 months.
Brief toxicological profile: Acute oral
toxicity (LD) for rats - 1369 - 2077 mg/kg; acute dermal
toxicity (LD) for rats - more than 2000 mg/kg; acute
inhalation toxicity (LC) for rats - 4.8 mg / cu. dm (4 hours).
Hygienic standards. MPC in water - 0.02 mg / cu. dm.
Scope of the drug. Clomazone is a selective herbicide used to control cereals and dicotyledonous weeds in soybean and rice crops during pre-emergence or pre-sowing application.
2. Method for determining clomazone in water
chromatographic methods
2.1. Key points
2.1.1. The principle of the technique
The technique is based on the extraction of clomazone from the analyzed sample with hexane, concentration of the extract and subsequent quantitative determination by alternative methods:
high performance liquid chromatography (HPLC) with
ultraviolet detector, gas liquid chromatography (GLC) with a constant recombination rate detector or thin layer chromatography (TLC). Quantitative determination is carried out by the method of absolute calibration.
2.1.2. Method selectivity
Under the proposed conditions, the method is specific in the presence of global environmental pollutants: chlorine derivatives of cycloparaffins (HCH isomers), diphenyl compounds (DDT and its derivatives), their metabolites - polychlorinated benzenes and phenols, as well as in the presence of sodium trichloroacetate, which can be used on crops in as a herbicide.
2.1.3. Metrological characteristic of the method (P = 0.95)
Reagents, solutions and materials
Clomazone with the content of d. 99.8%
(FMS, USA)
Nitrogen, och GOST 9293-79
Water ammonia, 25%, h GOST 1277-81
Acetone, h GOST 2603-79
n-Hexane, h GOST 2603-79
Hydrogen peroxide, 30% aqueous solution GOST 10929-77
Isopropyl alcohol, chemically pure TU 6-09-402-75
Sulfuric acid, chemically pure GOST 4203-77
Hydrochloric acid (hydrochloric), chemically pure GOST 3118-77
Methyl alcohol, chemically pure GOST
Sodium hydroxide, chemically pure, 25% aqueous solution GOST 4323-77
Sodium sulfate anhydrous, chemically pure GOST 1277-81
Silver nitrate, chemically pure GOST 1277-81
2-Phenoxymethanol, h TU 6-09-3688-76
Chromaton N-AW-DMCS (0.16 - 0.20 mm)
with 5% SE-30, Hemapol, Czech Republic
Chromaton N-AW-DMCS (0.16 - 0.20 mm) with 1.5
OV-17 + 1.95% QF-1, Hemapol, Czech Republic
Plates for HPTLC (USSR)
Records "Kieselgel 60 F-254" (Germany)
Records "Silufol" Czech Republic
Paper filters "white tape", ashless and pre-washed with hexane TU 6-09-2678-77
2.3. Cutlery, equipment, utensils
Liquid chromatograph Milichrome
with UV detector
Chromatographic steel column,
length 64 mm, inner diameter 2 mm,
filled with Silasorb 600, grain size 5 µm
Gas chromatograph series "Color" or
similar, equipped with a constant detector
recombination rate (RPR) with a limit
detection by lindane 4 x 10 g/cu. cm
Chromatographic glass column, length
1 or 2 m, inner diameter 2 - 3 mm
Microsyringe type MSH-10, capacity 10 µl TU 5E2-833-024
Apparatus for shaking type AVU-6s TU 64-1-2851-78
Water bath TU 64-1-2850-76
Analytical balance type VLA-200 GOST 34104-80E
Chromatographic chamber GOST 10565-74
Water jet pump GOST 10696-75
Mercury-quartz irradiator type OKN-11 TU 64-1-1618-77
Spray guns glass GOST 10391-74
Rotary vacuum evaporator IR-1M
or similar TU 25-11-917-76
Compressor unit TU 64-1-2985-78
Drying cabinet TU 64-1-1411-76E
Dividing funnels GOST 3613-75
Volumetric flasks, with a capacity of 100 ml GOST 1770-74
Measuring cylinders, with a capacity of 10, 50 ml GOST 1770-74E
Pear-shaped flasks with a thin section,
with a capacity of 100 ml GOST 10394-72
Flasks conical, with a capacity of 100 ml GOST 22524-77
Centrifuge test tubes, measured GOST 25336-82E
Pipettes, with a capacity of 0.1, 1, 2, 5 and 10 ml GOST 20292-74
Chemical funnels, conical, diameter
34 - 40 mm GOST 25336-82E
2.4. Sample selection
Sampling, storage and preparation of samples are carried out in accordance with
"Unified rules for sampling agricultural products, food products and environmental objects for the determination of trace amounts of pesticides", approved for N 2051-79 of 21.08.79
Samples taken can be stored in the refrigerator for up to 5 days. Before analysis, water (in the presence of suspension) is filtered through a loose paper filter.
2.5. Preparation for definition
2.5.1. HPLC method
2.5.1.1. Mobile phase preparation for HPLC
In a volumetric flask with a capacity of 100 ml, 5 ml of isopopanol and 5 ml of methanol are placed with a pipette, topped up to the mark with hexane, mixed, filtered.
2.5.1.2. Column conditioning
Rinse the HPLC column with hexane-methanol-isopropanol (90:5:5, v/v) for 30 min. at a solvent feed rate of 100 µl/min.
2.5.2. GLC method. Column preparation and conditioning
The finished packing (5% SE-30 on Chromaton N-AW-DMCS) is poured into a glass column, compacted under vacuum, the column is installed in the chromatograph thermostat without connecting to the detector, and stabilized in a nitrogen stream at a temperature of 250 -C for 10 - 12 noon
2.5.3. TLC method
2.5.3.1. Preparation of developing reagents
2.5.3.1.1. Developing reagent No. 1
1 g of silver nitrate is dissolved in 1 ml of distilled water, 10 ml of 2-phenoxymethanol, 190 ml of acetone, 1-2 drops of hydrogen peroxide are added, the solution is stirred and transferred into a dark glass bottle.
2.5.3.2.2. Developing reagent N 2
0.5 g of silver nitrate is dissolved in 5 ml of distilled water in a 100 ml volumetric flask, 10 ml of 25% aqueous ammonia is added, the solution is adjusted to 100 ml with acetone, mixed and transferred to a dark glass bottle.
2.5.3.2. Mobile phase preparation for TLC
In a volumetric flask with a capacity of 100 ml add 20 ml of acetone and add hexane to the mark, mix. The mixture is poured into the chromatographic chamber with a layer of no more than 6 - 8 mm in 30 minutes. Before starting chromatography.
2.5.4. Preparation of standard solutions
A 100 µg/mL clomazone stock standard solution is prepared by dissolving 0.010 g of a preparation containing 99.8% AI in hexane in a 100 mL volumetric flask. The solution is stored in the refrigerator for a month.
Working standard solutions with a concentration of 0.4; 1.0; 2.0; 4.0; 10.0; 20 and 40.0 µg/ml are prepared from clomazone stock standard solution by appropriate serial dilutions with hexane.
Working solutions are stored in the refrigerator for no more than a month.
2.5.5. Construction of a calibration graph
2.5.5.1. Calibration curve A (measurement according to paragraph 2.7.1, HPLC)
To build a calibration graph, 5 µl of a working standard solution of clomazone with a concentration of 4.0 is injected into the chromatograph injector; 10.0; 20.0 and 40 µg/ml.
2.5.5.2. Calibration curve B (measurement according to paragraph 2.7.2, GLC)
To build a calibration graph, 5 µl of a working standard solution of clomazone with a concentration of 0.4 is injected into the chromatograph evaporator; 1.0; 2.0; 4.0 and 10.0.
Carry out at least 5 parallel measurements. Find the average height of the chromatographic peak for each concentration. Build a calibration graph (A or B) of the dependence of the height of the chromatographic peak in mm on the concentration of clomazone in solution in µg/ml.
2.6. Definition Description
100 ml of the analyzed water sample is placed in a separating funnel with a capacity of 250 ml, 10 ml of a 25% aqueous solution of sodium hydroxide is added, mixed and 20 ml of n-hexane are added. The funnel is shaken for 3 minutes, after phase separation, the hexane layer is poured into a pear-shaped flask with a capacity of 100 ml, passing it through a layer of anhydrous sodium sulfate placed in a conical funnel on a pleated filter paper. The extraction of the drug from the aqueous sample is repeated twice more using 20 ml of n-hexane. The combined hexane extract is evaporated on a rotary vacuum evaporator at a temperature of 40 -C almost to dryness, the residue is blown off with a stream of air or nitrogen of special purity. The dry residue is dissolved in 0.1 (HPLC, TLC) or 0.25 ml (GLC) n-hexane and analyzed by one of the chromatographic methods.
2.7. Chromatography conditions
Liquid chromatograph with ultraviolet detector Milichrom (Russia).
Steel column 64 mm long, inner diameter 2 mm,
filled with Silasorb 600, grain size 5 microns.
Column temperature: room.
Mobile phase: hexane-isopropanol-methanol (90:5:5, v/v).
Eluent flow rate: 100 µl/min.
Operating wavelength: 240 nm.
Sensitivity: 0.4 units absorption on the scale.
Injection volume: 5 µl.
Clomazone exit time: about 6 min.
Linear detection range: 20 - 200 ng.
Samples producing peaks greater than the 40 µg/mL standard solution are diluted with HPLC mobile phase.
Gas chromatograph "Tsvet-570" with detector of constant ion recombination rate.
Glass column 1 m long, 3 mm inner diameter, filled with Chromaton N-AW-DMCS with 5% SE-30 (0.16 - 0.20 mm).
The working scale of the electrometer is 64 x 10 10 Ohm.
Recorder tape speed 200 mm/h.
Column thermostat temperature - 190 -С
detector - 300 -C
evaporator - 220 -C
The speed of the carrier gas (nitrogen) - 60 ml / min.
The volume of the injected sample is 5 µl.
The exit time of clomazone is 2.5 minutes.
Linear detection range: 2 - 50 ng.
Samples that produce peaks greater than the 10 µg/mL standard solution are diluted with hexane.
To improve the accuracy of clomazone identification in the presence of gamma-HCCH having a close retention time in the sample, clomazone is removed from the sample by treatment with concentrated sulfuric acid. Re-analysis of the sample allows you to establish the contribution of clomazone to the primary chromatographic signal.
Hexane solution in a flask, obtained according to paragraph 2.6 quantitatively
(or an aliquot thereof) is applied to chromatographic plates "Silufol", "Kieselgel 60F-254" or "Plates for HPTLC". Nearby, standard solutions are applied in a volume corresponding to the content of clomazone 1, 2, 5 and 10 μg. The plate is placed in a chromatographic chamber containing a mixture of n-hexane-acetone (4:1, v/v). After the development of the chromatogram, the plate is removed from the chamber, placed under draft until the solvents evaporate, then treated with one of the developing reagents and placed under an ultraviolet lamp for 5 minutes. The localization zone of the drug on the plates "Silufol", "Plates for HPTLC" and "Kieselgel 60F-254" appears as gray-brown spots with an Rf value of 0.35, 0.85 and 0.43, respectively. To determine clomazone by TLC, you can use plates "Alugram" and "Polygram" (manufactured by Germany). The Rf value of clomazone on these plates is 0.37 and 0.38, respectively.
3. Safety requirements
It is necessary to follow generally accepted safety rules when working with organic solvents, toxic substances, electric heaters.
4. Measurement error control
Operational control of the error and reproducibility of measurements is carried out in accordance with the recommendations of MI 2335-95. GSI "Internal quality control of the results of quantitative chemical analysis".
5. Developers
Yudina T.V., Fedorova N.E. (FNTSG named after F.F. Erisman).
Davidyuk E.I. (UkrNIIGINTOX, Kiev); Kisenko M.A., Demchenko V.F. (Institute of Occupational Medicine of the Academy of Sciences and the Academy of Medical Sciences of Ukraine, Kiev).
Liquid adsorption chromatography on a column
The separation of a mixture of substances in an adsorption column occurs as a result of their difference in sorbability on a given adsorbent (in accordance with the law of adsorption substitution established by M. S. Tsvet).
Adsorbents are porous bodies with a highly developed inner surface that hold liquids with the help of intermolecular and surface phenomena. These can be polar and non-polar inorganic and organic compounds. Polar adsorbents include silica gel (dried gelatinous silicon dioxide), aluminum oxide, calcium carbonate, cellulose, starch, etc. Non-polar sorbents - activated carbon, rubber powder and many others obtained synthetically.
The adsorbents are subject to the following requirements: S they must not enter into chemical reactions with the mobile phase and the substances to be separated; S must have mechanical strength; S grains of the adsorbent must be of the same degree of dispersion.
When choosing the conditions for the chromatographic process, the properties of the adsorbent and adsorbed substances are taken into account.
In the classical version of liquid column chromatography (LCC), an eluent (PF) is passed through a chromatographic column, which is a glass tube 0.5–5 cm in diameter and 20–100 cm long, filled with a sorbent (NP). The eluent moves under the influence of gravity. The speed of its movement can be adjusted by the crane at the bottom of the column. The mixture to be analyzed is placed at the top of the column. As the sample moves through the column, the components separate. At certain intervals, fractions of the eluent released from the column are taken, which are analyzed by any method that allows measuring the concentrations of analytes.
Column adsorption chromatography is currently used mainly not as an independent method of analysis, but as a method of preliminary (sometimes final) separation of complex mixtures into simpler ones, i.e. to prepare for analysis by other methods (including chromatographic). For example, a mixture of tocopherols is separated on an alumina column, the eluent is passed, and the α-tocopherol fraction is collected for subsequent photometric determination.
Chromatographic separation of the mixture on the column due to the slow advance of the PF takes a long time. To speed up the process, chromatography is carried out under pressure. This method is called High Performance Liquid Chromatography (HPLC)
The modernization of the equipment used in classical liquid column chromatography has made it one of the most promising and modern methods of analysis. High performance liquid chromatography is a convenient method for the separation, preparative isolation, and qualitative and quantitative analysis of non-volatile, thermolabile compounds of both low and high molecular weight.
Depending on the type of sorbent used, this method uses 2 chromatography options: on a polar sorbent using a non-polar eluent (direct phase option) and on a non-polar sorbent using a polar eluent - the so-called reversed-phase high-performance liquid chromatography (RP HPLC).
When the eluent passes to the eluent, the equilibrium under RPHLC conditions is established many times faster than under the conditions of polar sorbents and non-aqueous PFs. As a result of this, as well as the convenience of working with water and water-alcohol eluents, RPHLC has now gained great popularity. Most HPLC analyzes are carried out using this method.
Instrumentation for HPLC
A set of modern equipment for HPLC, as a rule, consists of two pumps 3,4 (Fig. 7.1.1.1), controlled by a microprocessor 5, and supplying eluent according to a specific program. Pumps create pressure up to 40 MPa. The sample is injected through a special device (injector) 7 directly into the eluent flow. After passing through the chromatographic column 8, the substances are detected by a highly sensitive flow detector 9, the signal of which is recorded and processed by the microcomputer 11. If necessary, fractions are automatically selected at the time of the peak output.
Columns for HPLC are made of stainless steel with an inner diameter of 2 - 6 mm and a length of 10-25 cm. The columns are filled with a sorbent (NF). Silica gel, alumina, or modified sorbents are used as NF. Silica gel is usually modified by chemically introducing various functional groups into its surface.
Detectors. Registration of the output from the column of a separate component is performed using a detector. For registration, you can use the change in any analytical signal coming from the mobile phase and related to the nature and amount of the mixture component. Liquid chromatography uses such analytical signals as light absorption or light emission of the exiting solution (photometric and fluorimetric detectors), refractive index (refractometric detectors), potential and electrical conductivity (electrochemical detectors), etc.
The continuously detected signal is recorded by the recorder. A chromatogram is a sequence of detector signals recorded on a tape recorder, which are generated when individual components of a mixture leave the column. In the case of separation of the mixture, separate peaks are visible on the external chromatogram. The position of the peak on the chromatogram is used for the purposes of identifying the substance, the height or area of the peak is used for the purposes of quantitative determination.
Qualitative Analysis
The most important characteristics of the chromatogram - the retention time tR and the retention volume associated with it - reflect the nature of the substances, their ability to sorption on the material of the stationary phase and, therefore, under constant chromatography conditions, they are a means of identifying the substance. For a given column with a certain flow rate and temperature, the retention time of each compound is constant (Fig. 7.1.1.2), where tR(A) is the retention time of component A of the analyzed mixture from the moment it is injected into the column until the maximum peak appears at the column outlet, 1K( ss) - retention time of the internal standard (substance initially absent in the analyzed mixture), h - peak height (mm), ab - peak width at half its height, mm.
To identify a substance by chromatogram, standard samples or pure substances are usually used. Compare the retention time of the unknown IR* component with the IRCT retention time of the known substances. But more reliable identification by measuring the relative retention time
In this case, a known substance (internal standard) is first introduced into the column and its retention time tR(Bc) is measured, then the test mixture is chromatographically separated (chromatographed), to which the internal standard is preliminarily added. The relative retention time is determined by formula (7.1.1.1).
Quantitative Analysis
This analysis is based on the dependence of the peak height h or its area S on the amount of substance. For narrow peaks, measurement h is preferable, for broad blurry peaks - S. The peak area is measured in different ways: by multiplying the peak height (h) by its width (ai / 2), measured at half its height (Fig. 7.2.3); planning; using an integrator. Modern chromatographs are equipped with electrical or electronic integrators.
Three methods are mainly used to determine the content of substances in a sample: the absolute calibration method, the internal normalization method, and the internal standard method.
The absolute calibration method is based on a preliminary determination of the relationship between the amount of the introduced substance and the area or height of the peak on the chromatogram. A known amount of the calibration mixture is introduced into the chromatogram and the areas or heights of the resulting peaks are determined. Build a graph of the area or height of the peak from the amount of injected substance. The test sample is analyzed, the area or height of the peak of the determined component is measured, and its amount is calculated based on the calibration curve.
This method provides information only on the relative content of the component in the mixture, but does not allow determining its absolute value.
The internal standard method is based on the comparison of a selected peak parameter of an analyte with the same parameter of a standard substance introduced into the sample in a known amount. A known amount of such a standard substance is introduced into the test sample, the peak of which is sufficiently well separated from the peaks of the components of the test mixture.
The last two methods require the introduction of correction factors that characterize the sensitivity of the detectors used to the analyzed substances. For different types of detectors and different substances, the sensitivity coefficient is determined experimentally.
Liquid adsorption chromatography also uses the analysis of fractions of solutions collected at the moment the substance exits the column. The analysis can be carried out by various physicochemical methods.
Liquid adsorption chromatography is used primarily for the separation of organic substances. This method is very successful in studying the composition of oil, hydrocarbons, effectively separating trans- and cis-isomers, alkaloids, etc. HPLC can be used to determine dyes, organic acids, amino acids, sugars, pesticide and herbicide impurities, medicinal substances and other contaminants in food products.
(mainly intermolecular) at the interface. As a method of analysis, HPLC is part of a group of methods, which, due to the complexity of the objects under study, includes the preliminary separation of the initial complex mixture into relatively simple ones. The simple mixtures obtained are then analyzed by conventional physicochemical methods or by special methods developed for chromatography.
The HPLC method is widely used in fields such as chemistry, petrochemistry, biology, biotechnology, medicine, food processing, environmental protection, drug production, and many others.
According to the mechanism of separation of the analyzed or separated substances, HPLC is divided into adsorption, distribution, ion-exchange, exclusion, ligand-exchange and others.
It should be borne in mind that in practical work, separation often proceeds not by one, but by several mechanisms simultaneously. So, exclusion separation can be complicated by adsorption effects, adsorption - distribution, and vice versa. At the same time, the greater the difference in substances in the sample in terms of the degree of ionization, basicity or acidity, in terms of molecular weight, polarizability, and other parameters, the more likely it is that a different separation mechanism will appear for such substances.
Normal phase HPLC
The stationary phase is more polar than the mobile phase, so the non-polar solvent predominates in the composition of the eluent:
- Hexane:isopropanol = 95:5 (for low polar substances)
- Chloroform:methanol = 95:5 (for medium polar substances)
- Chloroform:methanol = 80:20 (for highly polar substances)
Reverse phase HPLC
The stationary phase is less polar than the mobile phase, so water is almost always present in the eluent. In this case, it is always possible to ensure complete dissolution of the BAS in the mobile phase, it is almost always possible to use UV detection, almost all mobile phases are mutually miscible, gradient elution can be used, the column can be quickly re-equilibrated, the column can be regenerated.
Common eluents for reverse phase HPLC are:
- Acetonitrile: water
- methanol: water
- Isopropanol: water
Matrices for HPLC
Matrices used in HPLC are inorganic compounds such as silica (silica gel) or alumina, or organic polymers such as polystyrene (crosslinked with divinylbenzene) or polymethacrylate. Silica gel is, of course, now generally accepted.
The main characteristics of the matrix:
- Particle size (µm);
- Internal pore size (Å, nm).
Obtaining silica gel for HPLC:
- Formation of microspheres of polysilicic acid;
- Drying silica gel particles;
- Air separation.
Sorbent particles:
- Regular (spherical): higher pressure resistance, higher cost;
- Non-spherical: lower pressure resistance.
Pore size in HPLC is one of the most important parameters. The smaller the pore size, the worse their permeability for the molecules of the eluted substances. And consequently, the worse the sorption capacity of sorbents. The larger the pores, the lower, firstly, the mechanical stability of the sorbent particles, and, secondly, the smaller the sorption surface, hence, the worse the efficiency.
Stationary phase grafts
Normal phase HPLC:
- Stationary phase grafted with propylnitrile (nitrile);
- Stationary phase with propylamine grafting (amine).
Reverse phase HPLC:
- Stationary phase with alkyl graft;
- Stationary phase with alkylsilyl graft.
End-capping - protection of non-grafted areas of the sorbent by additional grafting with "small" molecules. Hydrophobic end-capping (C1, C2): higher selectivity, worse wettability; hydrophilic end-capping (diol): lower selectivity, higher wettability.
HPLC detectors
- UV
- diode array
- Fluorescent
- Electrochemical
- Refractometric
- mass selective
Links
Wikimedia Foundation. 2010 .
See what "High Performance Liquid Chromatography" is in other dictionaries:
high performance liquid chromatography- - [A.S. Goldberg. English Russian Energy Dictionary. 2006] Topics energy in general EN high performance liquid chromatographyHPLC … Technical Translator's Handbook
Term high performance liquid chromatography English term high performance liquid chromatography Synonyms Abbreviations HPLC, HPLC Related terms adsorption, oligopeptide, proteomics, sorbent, fullerene, endohedral, chromatography… …
Liquid chromatography, in order to increase the efficiency of separation, the solvent (eluent) under pressure (more than 3x107 Pa) is pumped through columns filled with a sorbent with particles of small diameter (up to 1 μm), and perfusion ... ...
A type of chromatography in which the liquid (eluent) serves as the mobile phase, and it is the stationary phase. sorbent, tv. a carrier with a liquid or gel applied to its surface. Carried out in a column filled with a sorbent (column chromatography), on a flat ... ... Natural science. encyclopedic Dictionary
- [κρώμα (υroma) color] a process based on the unequal ability of the individual components of the mixture (liquid or gaseous) to remain on the surface of the adsorbent both when they are absorbed from the carrier stream, and when ... ... Geological Encyclopedia
- (from other Greek ... Wikipedia
Term chromatography English term chromatography Synonyms Abbreviations Associated terms high performance liquid chromatography, clathrate, laboratory on a chip, porosimetry, proteome, proteomics, sorbent, enzyme, fullerene, endohedral… … Encyclopedic Dictionary of Nanotechnology
Liquid chromatography based on decomp. the ability of the separated ions to ion exchange with fixed. sorbent ions formed as a result of the dissociation of the ionogenic groups of the latter. Cation exchangers are used to separate cations, for ... ... Chemical Encyclopedia
HPLC- high performance liquid chromatography ... Dictionary of abbreviations of the Russian language
High performance liquid chromatography (HPLC) is one of the effective methods for separating complex mixtures of substances, which is widely used both in analytical chemistry and in chemical technology. The basis of chromatographic separation is participation ... Wikipedia
Books
- Practical High Performance Liquid Chromatography, Veronica R. Mayer. We present to the reader the 5th edition of the book, which has been expanded with modern methods and equipment. Much has been improved in the book and a large number of references have been added. The places in the text where...
9885 0
HPLC is liquid column chromatography, in which a variety of sorption mechanisms can be used. Essentially, HPLC is a modern form of classical liquid column chromatography. Listed below are some of the most significant qualitative characteristics of HPFA:
- high speed of the process, which made it possible to reduce the duration of separation from several hours and days to minutes;
- the minimum degree of blurring of the chromatographic zones, which makes it possible to separate compounds that differ only slightly in sorption constants;
- a high degree of mechanization and automation of information separation and processing, thanks to which column liquid chromatography has reached a new level of reproducibility and accuracy.
Intensive studies of the last decades, a huge amount of accumulated experimental data make it possible today to speak about the classification of variants within the framework of the high-performance liquid chromatography method. Of course, in this case, the classification according to the sorption mechanism given above remains valid.
A common classification is based on the comparative polarity of the mobile and stationary phases. A distinction is made between normal and reverse phase chromatography.
Normal-phase chromatography (NPC) is a variant of HPLC when the mobile phase is less polar than the stationary phase, and there is reason to believe that the main factor determining retention is the interaction of sorbates directly with the surface or volume of the sorbent.
Reversed-phase chromatography (RPC) is a variant of HPLC when the mobile phase is more polar than the stationary one, and retention is determined by direct contact of sorbate molecules with the surface or volume of the sorbent; in this case, ionized sorbates are not exchanged for ions of the mobile phase adsorbed on the surface.
Ion-exchange chromatography - a variant in which sorption is carried out by exchanging sorbed ions of the mobile phase for ions of chromatographed substances; ligand-exchange chromatography can be determined in exactly the same way.
Chromatography on dynamically modified sorbents is a variant of HPLC, in which the sorbate does not interact directly with the surface of the sorbent, but enters into association with the molecules of the near-surface layers of the eluent.
Ion-pair chromatography is a variant of reverse-phase chromatography of ionized compounds, in which a hydrophobic counterion is added to the mobile phase, which qualitatively changes the sorption characteristics of the system.
Size exclusion chromatography is a method for separating compounds according to their molecular weights, based on the difference in the diffusion rate in the pores of the stationary phase of molecules of various sizes.
For HPFA, a very important characteristic is the size of sorbents, usually 3-5 µm, now up to 1.8 µm. This makes it possible to separate complex mixtures of substances quickly and completely (the average analysis time is from 3 to 30 min).
The problem of separation is solved using a chromatographic column, which is a tube filled with a sorbent. During the analysis, a liquid (eluent) of a certain composition is fed through a chromatographic column at a constant speed. An accurately measured sample dose is injected into this stream. The components of the sample introduced into the chromatographic column, due to their different affinity to the sorbent of the column, move along it at different speeds and reach the detector sequentially at different times.
Thus, the chromatographic column is responsible for the selectivity and separation efficiency of the components. By selecting different types of columns, you can control the degree of separation of the analyzed substances. Compounds are identified by their retention time. The quantitative determination of each of the components is calculated based on the magnitude of the analytical signal measured using a detector connected to the output of the chromatographic column.
Sorbents. The formation of HPLC is largely associated with the creation of new generations of sorbents with good kinetic properties and various thermodynamic properties. The main material for sorbents in HPLC is silica gel. It is mechanically strong and has significant porosity, which gives a large exchange capacity for small column sizes. The most common particle size is 5-10 microns. The closer to the spherical shape of the particles, the lower the flow resistance, the higher the efficiency, especially if a very narrow fraction is screened out (for example, 7 +1 microns).
The specific surface area of silica gel is 10-600 m/g. Silica gel can be modified with various chemical groups grafted onto the surface (C-18, CN, NH2, SO3H), which makes it possible to use sorbents based on it to separate various classes of compounds. The main disadvantage of silica gel is its low chemical resistance at pH< 2 и рН >9 (silica dissolves in alkalis and acids). Therefore, an intensive search is currently underway for sorbents based on polymers that are stable at pH from 1 to 14, for example, based on polymethyl methacrylate, polystyrene, etc.
Sorbents for ion-exchange chromatography. Due to the peculiarities of separation (in an acidic or alkaline medium), the main material is sorbent-to-polystyrene with divinylbenzene of various degrees of cross-linking with SO3 -H + groups grafted to their surface (strongly acid cation exchangers) or -COO-Naf (weakly acid cation exchangers), -H2N + (CH3) 3Cl- (strong base anion exchangers) or -N+HR2Cl- (weak base anion exchangers).
Sorbents for gel-penetrating chromatography. The main type is styrene-DVB. Macroporous glasses, methyl methacrylate, silica gel are also used. For ion-exclusion chromatography, the same sorbents are used.
Pumps. To ensure the flow of the mobile phase (MP) through the column with the specified parameters, high pressure pumps are used. The most important specifications for LC pumps are: flow range; maximum working pressure; flow reproducibility; solvent supply pulsation range.
By the nature of the solvent supply, pumps can be of constant supply (flow) and constant pressure. Basically, in analytical work, a constant flow mode is used, when filling columns, a constant pressure mode is used. According to the principle of operation, pumps are divided into syringe pumps and plunger reciprocating pumps.
syringe pumps. Pumps of this type are characterized by the almost complete absence of pulsations in the flow of the mobile phase during operation. Disadvantages of the pump: a) high consumption of time and solvent for flushing when changing the solvent; b) suspension of separation during filling of the pump; c) large dimensions and weight while providing high flow and pressure (you need a powerful engine and a large piston force with its large area).
Plunger reciprocating pumps. Pumps of this type provide a constant volumetric flow of the mobile phase for a long time. Maximum working pressure 300-500 atm, flow rate 0.01-10 ml/min. Reproducibility of volumetric feed - 0.5%. The main drawback is that the solvent is fed into the system in the form of a series of successive pulses, so there are pressure and flow pulsations.
This is the main reason for the increased noise and desensitization of almost all detectors used in LC, especially electrochemical. Ways to combat pulsations: using double pumps or a two-plunger Bag-lai pump, using damping devices and electronic devices.
The amount of volumetric feed is determined by three parameters: the diameter of the plunger (usually 3.13; 5.0; 7.0 mm), its amplitude (12-18 mm) and frequency (which depends on the speed of rotation of the engine and gearbox).
Dosers. The purpose of the dispenser is to transfer a sample at atmospheric pressure to the inlet of a column at pressures up to several atmospheres. It is important that the dispenser is free of “dead” volumes that cannot be washed out by the mobile phase and that the sample does not wash out during dispensing. At first, dispensers in LC were similar to gas dispensers with a membrane puncture. However, the membranes do not hold more than 50-100 atm, their chemical resistance is insufficient, their pieces contaminate the column filters and capillaries.
In the liquid phase, the diffusion rate is much lower than in the gas phase. Therefore, it is possible to dispense with a flow stop - the sample does not have time to blur in the dispenser. During the injection of the sample into the dispenser, a special tap shuts off the flow of the solvent. The pressure at the inlet to the column quickly decreases, after a few seconds the sample can be injected into the dosing chamber with a conventional microsyringe. Next, the dispenser is locked, the solvent flow is turned on, and separation takes place.
The pressure that this valve holds is up to 500-800 atm. But when the flow stops, the equilibrium in the column is disturbed, which can lead to the appearance of “vacant” additional peaks.
Loop dispensers are the most widely used. When filling the dispenser under high pressure, there are inlets 1,2 and a channel between them. Inlets 3-6, the channels between them and the dosing loop are at atmospheric pressure, which allows you to fill the loop with a syringe or pump. As the dispenser is rotated, the flow of the mobile phase forces the sample into the column. To reduce the error, the loop is washed with 5-10 times the sample volume. If the sample is small, then it can be injected into the loop with a microsyringe. Loop volume is usually 5-50 μl.
ON THE. Voinov, T.G. Volova
Liquid chromatography This is a method for separating and analyzing complex mixtures of substances in which liquid is the mobile phase. It is applicable to the separation of a wider range of substances than the gas chromatography method. This is due to the fact that most substances are not volatile, many of them are unstable at high temperatures (especially high-molecular compounds) and decompose when converted to a gaseous state. The separation of substances by liquid chromatography is most often carried out at room temperature.
The features of all types of liquid chromatography are due to the fact that the mobile phase in it is a liquid, and the sorption of components from a gaseous and liquid eluent proceeds differently. If in gas chromatography the carrier gas performs only a transport function and is not sorbed by the stationary phase, then the liquid mobile phase in liquid chromatography is an active eluent, its molecules can be sorbed by the stationary phase. When passing through the column, the molecules of the components of the analyzed mixture that are in the eluent must displace the molecules of the eluent from the surface layer of the sorbent, which leads to a decrease in the energy of interaction between the molecules of the analyzed substance and the surface of the sorbent. Therefore, the values of retained volumes V R, proportional to the change in the free energy of the system, is smaller in liquid chromatography than in gas chromatography, and the range of linearity of the sorption isotherm in liquid chromatography is wider.
By using various eluents, one can change the retention parameters and selectivity of the chromatographic system. Selectivity in liquid chromatography, in contrast to gas chromatography, is determined not by one, but by two factors - the nature of the mobile (eluent) and stationary phases.
The liquid mobile phase has a higher density and viscosity than the gaseous phase, diffusion coefficients D well 3–4 orders of magnitude lower than in gas. This leads to a slowdown in mass transfer in liquid chromatography compared to gas chromatography. The Van Deemter equation due to the fact that the term V does not play a role in liquid chromatography ( D well D G), the graphic dependence of the efficiency also changes H on the linear velocity of the flow of the mobile phase has the form shown in fig. 1.9. 

In the classical version of column liquid chromatography, a glass column 1–2 m high, filled with a sorbent with a particle size of 100 μm and eluent, is injected with the analyzed sample dissolved in the eluent, and the eluent is passed through, taking portions of the eluate at the outlet of the column. This variant of liquid chromatography is still used in laboratory practice, but since the eluent flow rate under the action of gravity is low, the analysis is lengthy.
The modern version of liquid chromatography, the so-called high-performance liquid chromatography HPLC, uses volumetric and surface-porous sorbents with a particle size of 5–10 μm, pressure pumps that provide pressure in the system up to 400 atm, and highly sensitive detectors. Fast mass transfer and high separation efficiency make it possible to use HPLC for the separation of molecules (liquid-adsorption and liquid-liquid partition chromatography), for the separation of ions (ion-exchange, ion, ion-pair chromatography), for the separation of macromolecules (size-exclusion chromatography).
1.3. SOLVENT RETENTION AND STRENGTH
In order for the analytes to be separated on the column, as mentioned above, the capacity factor k" must be greater than 0, i.e. the substances must be retained by the stationary phase, the sorbent. However, the capacity factor should not be too large to obtain an acceptable elution time. If a stationary phase is chosen for a given mixture of substances, which holds them, then further work on the development of an analysis procedure consists in choosing a solvent that would provide, in the ideal case, different for all components, but acceptable not very large k. This is achieved by changing the eluting strength of the solvent.

In the case of adsorption chromatography on silica gel or alumina, as a rule, the strength of a two-component solvent (for example, hexane with the addition of isopropanol) is increased by increasing the content of the polar component (isopropanol) in it, or reduced by decreasing the content of isopropanol. If the content of the polar component becomes too low (less than 0.1%), it should be replaced with a weaker eluting strength. The same is done, replacing either the polar or non-polar component with others, even if this system does not provide the desired selectivity with respect to the mixture components of interest. When selecting solvent systems, both the solubilities of the mixture components and the eluotropic series of solvents compiled by different authors are taken into account.
Approximately in the same way, the strength of the solvent is selected in the case of using grafted polar phases (nitrile, amino, diol, nitro, etc.), taking into account possible chemical reactions and excluding solvents dangerous for the phase (for example, aldehydes and ketones for the amino phase).
In the case of reverse phase chromatography, the strength of the solvent is increased by increasing the content of the organic component (methanol, acetonitrile or THF) in the eluent and reduced by adding more water. If the desired selectivity cannot be achieved, another organic component is used or attempts are made to change it with the help of various additives (acids, ion-pair reagents, etc.).
In separations by ion-exchange chromatography, the strength of the solvent is changed by increasing or decreasing the concentration of the buffer solution or by changing the pH, in some cases modification with organic substances is used.
However, especially in the case of complex natural and biological mixtures, it is often not possible to choose the solvent strength in such a way that all sample components are eluted within an acceptable time. Then one has to resort to gradient elution, i.e. use a solvent whose eluting strength during the analysis changes so that it constantly increases according to a predetermined program. With this technique, it is possible to achieve the elution of all components of complex mixtures in a relatively short period of time and their separation into components in the form of narrow peaks.
1.6.1. Adsorption liquid chromatography. Adsorption liquid chromatography, depending on the polarity of the stationary and mobile phases, is divided into normal phase (NPC) and reversed phase (RPC) chromatography. In NPC, a polar adsorbent and non-polar mobile phases are used; in OFC, a non-polar adsorbent and polar mobile phases are used, but in both cases, the choice of the mobile phase is often more important than the choice of the stationary one. The stationary phase must hold the substances to be separated. The mobile phase, i.e. the solvent, must provide different column capacities and efficient separations in a reasonable time.

As a stationary phase in adsorption liquid chromatography, polar and non-polar finely dispersed porous materials with a specific surface area of more than 50 m 2 /g are used. Polar adsorbents (SiO 2 ,Al 2 O 3 , florisil, etc.) have weakly acidic groups on the surface, capable of retaining substances with basic properties. These adsorbents are mainly used for the separation of non-polar and medium polar compounds. Their drawback is high sensitivity to the water content in the eluents used. To eliminate this drawback, polar sorbents are treated with amines, diols, and other reagents, resulting in surface grafting of these reagents, surface modification, and a change in selectivity with respect to the analytes.
Non-polar adsorbents (graphitized soot, diatomite, kieselguhr) are non-selective to polar molecules. To obtain non-polar adsorbents, non-polar groups are often grafted onto the surface, for example, of silica gel, for example, alkylsilyl - SiR 3, where R - alkyl groups are C 2 - C 22.
The mobile phase should completely dissolve the analyzed sample, have a low viscosity (so that the diffusion coefficients are large enough), it is desirable that it is possible to separate the separated components from it. It must be inert to the materials of all parts of the chromatograph, safe, cheap, suitable for the detector.
The mobile phases used in liquid chromatography differ in their eluting strength. The eluting power of a solvent shows how many times the sorption energy of a given eluent on a given adsorbent is greater than the sorption energy of the eluent chosen as a standard, for example, n-heptane. Weak solvents are poorly adsorbed by the stationary phase, so the distribution coefficients of sorbed substances (sorbate) are high. Conversely, strong solvents adsorb well, so the sorbate partition coefficients are low. The stronger the solvent, the higher the solubility of the analyzed sample in it, the stronger the solvent-sorbate interaction.
To ensure high separation efficiency on the column, it is necessary to select a mobile phase that has the optimal polarity for the mixture to be separated under the selected separation conditions. Typically, the stationary phase is selected first, which has a polarity close to that of the components to be separated. Then the mobile phase is selected, ensuring that the capacitance coefficient k" turned out to be in the range from 2 to 5. If the polarity of the mobile phase is too close to the polarity of the stationary phase, the retention time of the components will be too short, and if the polarities of the mobile and stationary phases differ very much, the retention time becomes too long.
When selecting mobile phases, they are guided by the so-called eluotropic series, based on the use of polarity indices Snider R", which subdivides all solvents into strong (polar) and weak (weakly polar and non-polar). The polarity scale is based on the solubility of substances used as mobile phases in dioxane, nitromethane and ethanol.
Table 1.2 shows the values of the polarity index and elution strength (with respect to SiO 2 ) of a number of solvents most commonly used in liquid chromatography as mobile phases. The short-wavelength transparency limits of these solvents are also indicated here, which facilitates the selection of conditions for detecting mixture components.
Table 1.2
Characteristics of solvents used in liquid chromatography
|
Solvent |
Polarity index |
Eluting power (SiO 2) |
Shortwave Transparency Limit |
|
Fluoralkane | |||
|
Cyclohexane | |||
|
n-Hexane | |||
|
Carbon tetrachloride | |||
|
Diisopropyl ether | |||
|
diethyl ether | |||
|
dichloromethane | |||
|
Tetrahydrofuran | |||
|
Chloroform | |||
|
Acetic acid | |||
|
Acetonitrile | |||
|
Nitromethane | |||
Liquid chromatography often uses not individual solvents, but their mixtures. Often, minor additions of another solvent, especially water, greatly increase the eluting strength of the eluent.
When separating multi-component mixtures, one mobile phase as eluent may not separate all sample components in a reasonable time. In this case, a stepwise or gradient elution method is used, in which increasingly stronger eluents are used sequentially during chromatography, which makes it possible to elute highly retained substances in less time.
In liquid chromatography, there are some empirical rules that are very useful when choosing an eluent:
sorption of a compound, as a rule, increases with an increase in the number of double bonds and OH groups in it;
sorption decreases in a number of organic compounds: acidsalcoholsaldehydesketonesestersunsaturated hydrocarbonssaturated hydrocarbons;
to separate substances of different polarity and separate substances of different classes, normal-phase chromatography is used: the analyzed sample is dissolved and eluted with a non-polar eluent, compounds of different classes leave the column with a polar adsorbent for different times, while the retention time of compounds with different functional groups increases during the transition from non-polar compounds to weakly polar ones. For very polar molecules, retention times are so long that analysis is not possible when using a non-polar eluent. To reduce the retention time of polar components, one passes to polar eluents;
in the reversed-phase variant, the stationary phase (non-polar adsorbent) adsorbs the non-polar component more strongly from polar eluents, for example, from water;
By reducing the polarity of the eluent by adding a less polar solvent, the retention of the components can be reduced.
1.6.2. Partition liquid chromatography. In partition or liquid-liquid chromatography, the separation of the components of the analyzed sample is due to differences in the coefficients of their distribution between two liquid phases that do not mix with each other, one of which is stationary and is located on the surface or in the pores of a solid immovable carrier, and the second is mobile.
In terms of the nature of the interaction forces that cause a different distribution between the two phases of substances that differ in their chemical structure, distribution chromatography is similar to adsorption chromatography, i.e., here the separation is also based on the difference in the forces of intermolecular interaction between the components of the analyzed sample with the stationary and mobile liquid phases.
Depending on the performance technique, partition chromatography, like adsorption chromatography, can be column or planar (paper or thin layer).
As solid carriers, substances are used that are indifferent to the mobile solvent and the components of the analyzed sample, but capable of retaining the stationary phase on the surface and in the pores. Most often, polar substances (cellulose, silica gel, starch) are used as carriers. They are applied to the stationary phase - a polar solvent, most often water or alcohol. In this case, less polar or non-polar substances (alcohols, amines, ketones, hydrocarbons, etc.) are used as mobile phases. This type of partition chromatography is called normal-phase. It is used to separate polar substances.
The second variant of partition chromatography differs in that non-polar carriers (rubber, PTFE, hydrophobized silica gel) are used as the stationary solid phase, non-polar solvents (hydrocarbons) as the stationary liquid phase, and polar solvents (alcohols, aldehydes) as the mobile liquid phase. , ketones, etc., often water). This variant of partition chromatography is called reverse phase and is used to separate non-polar substances.
To achieve optimal separation of the components of the analyzed sample, the selection of the mobile phase is very important. Solvents (mobile and stationary liquid phases) should be selected so that the distribution coefficients of the mixture components differ significantly. The liquid phases are subject to the following requirements:
1) the solvents used should dissolve the substances to be separated well, and their solubility in the stationary phase should be greater than in the mobile one;
2) the solvents used as mobile and stationary phases must be mutually saturated, i.e. the composition of the solvent must be constant during passage through the column;
3) the interaction of solvents used as the mobile phase with the stationary phase should be minimal.

Most often, in partition liquid chromatography, not individual substances are used as mobile liquid phases, but their mixtures in various ratios. This makes it possible to regulate the polarity of the mobile phase, change the ratio of the polarities of the mobile and stationary phases, and achieve optimal conditions for separating the components of a particular mixture being analyzed.
A significant disadvantage of this chromatographic method is the rather rapid washing off of the deposited stationary liquid phase from the carrier. To eliminate it, the solvent used as the mobile phase is saturated with the substance used as the stationary liquid phase, or the stationary liquid phase is stabilized by grafting it onto a carrier.
A variation of partition liquid chromatography is the widely used HPLC method.

The most common chromatographic systems are systems that have a modular assembly principle. Pumps, degassers, detectors, dispensers (autosamplers), column ovens, fraction collectors, chromatography system controls and recorders are available as separate modules. A wide range of modules allows you to flexibly solve various analytical problems, quickly change the system configuration if necessary, with minimal costs. At the same time, monomodular (integrated) LCs are also produced, the main advantage of which is the miniaturization of individual blocks and the compactness of the device.
Depending on the method of elution, liquid chromatographs are divided into isocratic and gradient.
Diagram of an isocratic chromatograph
The mobile phase from the tank (1) through the inlet filter (9) is supplied by a high-pressure precision pump (2) to the sample input system (3) - a manual injector or an autosampler, where the sample is also injected. Further, through the in-line filter (8), the sample with the current of the mobile phase enters the separation element (elements) (4) - through the pre-column into the separation column. Then, the eluate enters the detector (5) and is removed into the drain tank (7). When the eluate flows through the measuring circuit of the detector, the chromatogram is registered and the data is transmitted to an analog recorder (recorder) (6) or another system for collecting and processing chromatographic data (integrator or computer). Depending on the design of the functional modules, the system can be controlled from the keyboard of the control module (usually a pump or system controller), from the keyboards of each of the system modules, or by a control program from a personal computer.
In the case of gradient elution, two fundamentally different types of liquid chromatographs are used. They differ in the point of formation of the mobile phase composition gradient.
Scheme of a gradient chromatograph with the formation of a gradient of the composition of the mobile phase on the low pressure line.
The mobile phase from the tanks (1) through the inlet filters (9) and the gradient programmer (10) is supplied by a high-pressure precision pump (2) to the sample injection system (3) - a manual injector or an autosampler, where the sample is also injected. The operation of the gradient programmer valves is controlled either by the system control module (pump or controller) or by a PC control program. Systems of this type form a binary, three-dimensional and four-dimensional gradient. The form of the gradient processing function depends on the specific control module or control program, as well as the functionality of the controlled and control modules. Further, through the in-line filter (8), the sample with the current of the mobile phase enters the separation element (elements) (4) - through the pre-column into the separation column. Then, the eluate enters the detector (5) and is removed to the drain tank (7). When the eluate flows through the measuring circuit of the detector, the chromatogram is registered and the data is transmitted to an analog recorder (recorder) (6) or another system for collecting and processing chromatographic data (integrator or computer). Depending on the design of the functional modules, the system can be controlled from the keyboard of the control module (usually a pump or system controller), or by a control program from a personal computer. In the case of controlling the control module, it is possible to independently control the detector from its own keyboard.
Despite the apparent attractiveness of such systems (they use only one high-pressure precision pump), these systems have a number of disadvantages, among which the main one is, perhaps, the severe need for thorough degassing of the mobile phase components even before the low-pressure mixer (gradient programmer chamber). It is carried out with the help of special flow degassers. Because of this fact, their cost becomes comparable to another type of gradient systems - systems with the formation of a gradient composition of the mobile phase on the high pressure line.
The fundamental difference between systems with the formation of a mobile phase gradient composition in the high pressure line is the mixing of components in the high pressure line, naturally, with this approach, the number of precision pumps is determined by the number of tanks for mixing the mobile phase. With this approach, the requirements for the thoroughness of the degassing of the components are significantly reduced.
Scheme of a gradient chromatograph with the formation of a gradient of the composition of the mobile phase on the high pressure line.
The mobile phase from the tanks (1) through the inlet filters (9) is supplied by precision high-pressure pumps (2 and 11) through a static or dynamic flow mixer (10) to the sample injection system (3) - a manual injector or an autosampler, where the sample is also injected. The operation of controlled pumps is controlled either by the system's control module (master pump or controller) or by a PC control program. In this case, all pumps are controlled. Systems of this type form a binary or three-dimensional gradient. The form of the gradient processing function depends on the specific control module or control program, as well as the functionality of the controlled and control modules. Further, through the in-line filter (8), the sample with the current of the mobile phase enters the separation element (elements) (4) - through the pre-column into the separation column. Then the eluate enters the detector (5) and is removed into the drain tank (7). When the eluate flows through the measuring circuit of the detector, the chromatogram is registered and the data is transmitted to an analog recorder (recorder) (6) or another system for collecting and processing chromatographic data (integrator or computer). Depending on the design of the functional modules, the system can be controlled from the keyboard of the control module (usually a pump or system controller), or by a control program from a personal computer. In the case of controlling the control module, it is possible to independently control the detector from its own keyboard.
The proposed schemes are rather simplified. Additional devices can be included in the systems - a column thermostat, post-column derivatization systems, sample preparation and concentration systems, a solvent recycler, membrane systems for suppressing background electrical conductivity (for ion chromatography), additional protective systems (filters, columns), etc. In the diagrams, the manometric modules are also not shown separately. As a rule, these devices are built into pump units. These units can combine multiple pumps, a pump with a gradient programmer, and a common system controller. The structure of the system depends on its configuration and each specific manufacturer.
Such a radical complication of the technical support of the chromatographic process leads to the emergence of a number of requirements for the properties of the mobile phase, which are absent in classical column and planar chromatography. The liquid phase must be suitable for detection (be transparent in a given region of the spectrum or have a low refractive index, a certain electrical conductivity or permittivity, etc.), inert to the materials of the parts of the chromatographic tract, not form gas bubbles in the pump valves and the detector cell, not have mechanical impurities.

In liquid chromatography many types of pumps are used. Low pressure LC often uses peristaltic pumps (Figure 1).
Fig.1 MasterFlex programmable peristaltic pump.
In HPLC, high pressure pumps are used to ensure the flow of the mobile phase through the column with the specified parameters.
The most important technical characteristics of HPLC pumps are: flow range; maximum working pressure; flow reproducibility; solvent supply pulsation range.
By the nature of the solvent supply, pumps can be of constant supply (flow) and constant pressure. Basically, in analytical work, a constant flow mode is used, when filling columns, a constant pressure mode is used.
According to the principle of operation, HPLC pumps are divided into syringe and on plunger reciprocating .
Syringe pumps
The main distinguishing feature of these pumps is their cyclical operation, and therefore the chromatographs in which these pumps are used also differ in cyclical operation.
Rice. 2. Principal arrangement of a syringe pump for HPLC.
Rice. 2A. Syringe pump.
The control unit BU supplies voltage to the motor D, which determines the speed and direction of its rotation. The rotation of the engine with the help of the gearbox P is converted into the movement of the piston P inside the cylinder D. The pump is operated in 2 cycles. In the filling cycle, valve K2 is closed, K1 is open, the solvent flows from the tank into cylinder C. In the supply mode, valve K1 is closed, and through valve K2 the mobile phase enters the dosing device.
Pumps of this type are characterized by the almost complete absence of pulsations in the flow of the mobile phase during operation.
Pump Disadvantages:
a) high consumption of time and solvent for washing when changing the solvent;
b) the volume of PF limited by the volume of the syringe, and hence the limited separation time;
c) suspension of separation during filling of the pump;
d) large dimensions and weight while providing high flow and pressure (you need a powerful engine and a large piston force with its large area).
Plunger reciprocating pumps.
Rice. 3. Principal device of a plunger pump.
Operating principle.
The engine D through the gearbox R reciprocates the plunger P, moving in the working head of the pump. Valves K1 and K2 open when the pump is in the suction and delivery phase respectively. The amount of volumetric feed is determined by three parameters: plunger diameter (usually 3.13; 5.0; 7.0 mm), its amplitude (12-18 mm) and frequency (which depends on the speed of rotation of the engine and gearbox).

Pumps of this type provide a constant volumetric flow of the mobile phase for a long time. Maximum working pressure 300-500 atm, flow rate 0.01-10 ml/min. Volume feed repeatability -0.5%. The main drawback is that the solvent is fed into the system in the form of a series of successive pulses, so there are pressure and flow pulsations (Fig. 4). This is the main reason for the increased noise and desensitization of almost all detectors used in LC, especially electrochemical.
Fig.4. Plunger pump pulsation.
Ways to deal with pulsations.
1. Application of damping devices.
These are spiral tubes of a special profile made of stainless steel, included in series or in parallel in the system between the pump and the dispenser.
Rice. 5. Spiral damper.
The damper is untwisted with an increase in pressure in it (acceleration of the pump). When the pressure drops, it twists, its volume decreases, it squeezes out part of the solvent, maintaining a constant flow rate and reducing pulsations. Such a damper works well at a pressure of 50 atm and above.
At a pressure of 5-30 atm, it smooths out pulsations better air damper, made from a column (Fig. 6.). The air in the plugged column (6x200 mm) is compressed and the pulsations are extinguished. The air in it dissolves in 24 hours.
Rice. 6. Air damper.
2. The use of electronic devices.
When using an electronic pressure transducer, the readings from the transducer can be used to control the operation of the pump. When the pressure drops, the engine speed increases and compensates for the decrease in pressure. It is also possible to compensate for leaks in the valves and partially in the cuffs. The use of an electronic damper (BPZh-80, KhPZh-1, etc.) makes it possible to reduce pressure pulsations to 1 atm at a pressure of 100-150 kgf/cm2.
1.6.3. Ion-exchange, ion, ion-pair chromatography. The methods of ion-exchange, ion and ion-pair chromatography are based on the dynamic process of replacing ions associated with the stationary phase with eluent ions entering the column. The main goal of the chromatographic process is the separation of inorganic or organic ions of the same sign. Retention in these types of chromatography is determined by the change in the free energy of the ion exchange reaction. The ratio of the concentrations of exchanging ions in the solution and in the sorbent phase is characterized by ion-exchange equilibrium. Ion exchange consists in the fact that some substances (ion exchangers), when immersed in an electrolyte solution, absorb cations or anions from it, releasing an equivalent amount of other ions with a charge of the same sign into the solution. Between the cation exchanger and the solution there is an exchange of cations, between the anion exchanger and the solution there is an exchange of anions.
Cation exchangers are most often specially synthesized insoluble polymeric substances containing acidic ionogenic groups in their structure: -SO 3 H; –COOH; -OH; –PO 3 H 2 ; –AsO 3 H 2 .
The chemical formulas of cation exchangers can be schematically represented as R-SO 3 H; R-SO 3 Na. In the first case, the cation exchanger is in the H-form, in the second, in the Na-form. R is a polymer matrix.
Cation exchange reactions are written as ordinary heterogeneous chemical reactions:
RN + Na + RNa + H +
Anion exchangers contain basic ionogenic groups in their structure: –N(CH 3) 3 + ; =NH 2 + ; =NH + etc. Their chemical formulas can be represented as RNH 3 OH and RNH 3 Cl or ROH, RCl. In the first case, the anion exchanger is in the OH form, in the second, in the Cl form. The anion exchange reaction can be written as follows:
R–OH+Cl – RCl+OH –
Amphoteric ion exchangers are known that contain both acidic and basic groups in their structure. Ion exchangers that have in their composition the same type (for example, SO 3 H) acidic (basic) groups are called monofunctional; ion exchangers containing heterogeneous (for example, - SO 3 H, - OH) acidic (basic) groups - polyfunctional.
Monofunctional ion exchangers are obtained by a polymerization reaction. The polycondensation reaction makes it possible to obtain polyfunctional ion exchangers. In order for the resulting ion exchangers to have sufficiently high performance characteristics, they must be insoluble, but swellable in the appropriate solvent and have a sufficiently large number of ionogenic groups capable of exchanging with the ionogenic groups of the analyzed sample. This can be achieved if the resulting polymer chains are sufficiently branched and connected to each other by "cross-linking bridges". For example, in the preparation of polymerization type cation exchangers based on styrene, divinylbenzene is most often used as a crosslinking agent, the introduction of which in an amount of up to 16% ensures the production of ion exchangers with various degrees of swelling and, therefore, allows one to control the ion exchanger porosity. The degree of swelling of the ion exchanger, expressed in milliliters/gram, is the volume of 1 g of air-dry ion exchanger packed into the column.
The ion exchanger absorbs, as a rule, one of the counterions - ions in the mobile phase, i.e. it exhibits a certain selectivity. Series of affinity, or selectivity, of ions with respect to ion exchangers of various types have been experimentally established. For example, at low solution concentrations on strongly acidic cation exchangers, ions with the same charge are sorbed in the following sequence:
Li + Na + K + Rb + Cs +
Mg 2+ Ca 2+ Sr 2+ Ba 2+ .
For ions with different charges, the sorbability increases with increasing charge:
Na+Ca2+ However, changing the conditions for carrying out the ion exchange reaction can lead to series inversion. Affinity series have also been established for anion exchangers. For example, the sorbability of anions on strongly basic anion exchangers increases in the series: F - OH - Cl - Br - NO 3 - J - SCN - ClO 4 - . Ion exchangers containing strongly acidic or strongly basic groups in their structure enter into ion exchange reactions with any ions in solution that have charges of the same sign as the sign of the counterion. Such ion exchangers are called universal. The process of ion exchange between the analyte and the ion exchanger can be carried out in one of three ways: static, dynamic (ion exchange filter method) and chromatographic. Static method
ion exchange is that a sample of the ion exchanger is brought into contact with a certain volume of solution and stirred or shaken for a certain time until equilibrium is established. This is a fast and simple method of ion exchange, used to concentrate ions from dilute solutions, remove unwanted impurities, but it does not provide complete absorption of ions, since ion exchange is a non-equilibrium process, and therefore does not guarantee complete separation of ions. When carrying out ion exchange in a dynamic way
a solution is passed through the column with an ion exchanger, which, as it moves along the column, comes into contact with new granules of the ion exchanger. This process provides a more complete exchange than the static method, since the exchange products are removed by the solution flow. They can concentrate ions from dilute solutions and separate ions that differ greatly in properties, for example, differently charged ions (separate cations from anions), but the separation of ions of the same charge sign is almost impossible. Quantitative separation of such ions is possible only with repeated repetition of sorption-desorption elementary acts under dynamic conditions, i.e. chromatographic method
. When working with this method, high layers of ion exchanger are used, and the mixture to be separated is introduced into this layer in an amount much less than the capacity of the column, due to which repeated repetition of elementary acts of ion exchange is ensured. In terms of the analysis technique, ion-exchange chromatography is similar to molecular chromatography and can be carried out according to the eluent (developing), frontal, and displacement options. The difference between molecular and ion-exchange chromatography is that in molecular chromatography, the separated components of the mixture are eluted from the column with a pure eluent, while in ion-exchange chromatography, an electrolyte solution is used as an eluent. In this case, the exchanged ion of the eluent should be sorbed less selectively than any of the ions of the mixture being separated. When carrying out developing ion-exchange chromatography, which is used most often, a column filled with an ion exchanger is first washed with an electrolyte solution until the ion exchanger completely replaces all its ions with the ions contained in the eluent. Then, a small volume of the analyte solution containing separable ions in an amount of about 1% of the capacity of the ion exchanger is injected into the column. Next, the column is washed with an eluent solution, taking fractions of the eluate and analyzing them. A mixture of Cl - , Br - , J - ions can be separated on a highly basic anion exchange resin (cross-linked polystyrene containing groups of quaternary ammonium bases N (CH 3) 3 +), for example, AB-17, which has a range of selectivity (selectivity): NO 3 - Cl – Br – J – . As a result, a NaNO 3 solution is used as an eluent. First, this solution is passed through the ion exchanger until it is completely saturated with NO 3 - ions. When the mixture to be separated is introduced into the column, ions Cl – , Br – , J – are absorbed by the anion exchanger, displacing NO 3 – ions. During subsequent washing of the column with NaNO 3 solution, the ions Cl – , Br – , J – in the upper layers of the anion exchanger are gradually again replaced by NO 3 – ions. Cl - ions will be displaced the fastest of all, J - ions will stay in the column the longest. The difference in the selectivity of the ion exchanger to the ions of the mixture leads to the fact that separate zones of adsorbed ions Cl – , Br – and J – are formed in the column, moving through the column at different speeds. As you move along the column, the distance between zones increases. In each zone there is only one of the anions of the mixture being separated and the anion of the eluent, in the interval between the zones there is only an anion of the eluent. Thus, fractions containing individual components of the mixture to be separated will appear in the eluent at the column outlet. To solve practical problems, the conditions for ion separation are varied by selecting a suitable mobile phase (composition, concentration, pH, ionic strength) or by changing the porosity of the polymer matrix of the ion exchanger, i.e., the number of interchain bonds in the matrix, and creating ion exchange sieves that are permeable to some ions and capable of their exchange and impenetrable to others. It is also possible to change the nature and mutual arrangement of ionogenic groups, as well as to obtain sorbents capable of selective chemical reactions due to complex formation. High selectivity is possessed, for example, by complexing ion exchangers containing in their structure chelating groups of organic reagents dimethylglyoxime, dithizone, 8-hydroxyquinoline, etc., as well as crown ethers. The greatest application in ion exchange, ion, and ion pair chromatography is found in synthetic macro- and micronet organic ion exchangers having a large exchange capacity (3–7 mmol/g), as well as inorganic ion-exchange materials. Micromesh ion exchangers are capable of exchanging ions only in the swollen state, while macromesh ones are capable of exchanging ions in the swollen and unswollen states. Another structural type of ion exchangers are surface-film ion exchangers, the solid core of which is made of a non-porous copolymer of styrene and divinylbenzene, glass or silica gel and is surrounded by a thin film of the ion exchanger. The total diameter of such a particle is about 40 µm, and the thickness of the ion exchanger film is 1 µm. The disadvantage of such ion exchangers is the relatively large particle diameter and low exchange capacity due to the low specific surface area, as a result of which it is necessary to work with small samples and, accordingly, to use highly sensitive detectors. In addition, such ion exchangers are quickly poisoned and are not capable of regeneration. In high-performance ion-exchange and ion chromatography, volumetric-porous polystyrene ion exchangers, volumetric-porous silicas with a granule diameter of about 10 μm, and practically non-swelling surface-porous and surface-modified copolymers of styrene and divinylbenzene with ionogenic sulfo and amino groups are used. In ion-pair chromatography, "brush" sorbents are used - silica gels with grafted reversed phases C 2, C 8, C 18, which are easily converted into a cation exchanger upon absorption of ionic surfactants from the mobile phase, for example, alkyl sulfates or salts of quaternary ammonium bases. When carrying out chromatographic separation using ion exchangers, aqueous solutions of salts are most often used as the mobile phase. This is due to the fact that water has excellent dissolving and ionizing properties, due to which the molecules of the analyzed sample instantly dissociate into ions, the ion exchange groups of the ion exchanger are hydrated and also go into a fully or partially dissociated form. This ensures a rapid exchange of counterions. The eluting strength of the mobile phase is mainly influenced by pH, ionic strength, the nature of the buffer solution, the content of the organic solvent or surfactant (ion-pair chromatography). The pH value is chosen depending on the nature of the ionogenic groups, the ions to be separated, and the matrix. It is possible to work with strongly acidic and strongly basic ion exchangers at pH = 2–12, with weakly acidic ones at pH = 5–12, and with weakly basic ones at pH = 2–6. Silica-based sorbents cannot be used at pH 9. The ionic strength of the mobile phase affects the capacity of the ion exchanger. With an increase in ionic strength, the sorption of ions usually decreases, since the eluting strength of the mobile phase increases. Therefore, at the beginning of separation, the mobile phase should have a low ionic strength (0.05–0.1), and the final value of this characteristic should not exceed 2. In gradient elution, buffers with increasing ionic strength are often used. For the selective elution of ions absorbed by the ion exchanger, you can use water, buffer solutions (phosphate, acetate, borate, hydrocarbonate, etc.) with a certain pH value and ionic strength, solutions of mineral (hydrochloric, nitrogen, sulfuric, phosphoric) and organic (phenol, citric, lactic, tartaric, oxalic, EDTA) acids. The choice of eluent is facilitated by the fact that the limiting distribution coefficients of most elements between aqueous (water-organic) solutions of many complexants and standard-type ion exchangers are determined and presented in tables. 1.6.4. Size exclusion chromatography. Size exclusion chromatography is a type of liquid chromatography in which the separation of components is based on the distribution of molecules according to their size between the solvent in the pores of the sorbent and the solvent flowing between its particles. During separation, small molecules enter the polymer network, in the pores of which the solvent serves as a stationary phase, and are retained there. Large molecules cannot penetrate the polymer network and are washed out of the column by the mobile phase. The largest molecules are eluted first, then the medium ones, and finally the small ones. Size exclusion chromatography is subdivided into gel permeation and gel filtration. In gel permeation chromatography, separation occurs on polymers that swell in organic solvents. The gel filtration version of size exclusion chromatography involves the use of water-swellable polymers as stationary phases. The duration of retention of the components of the analyzed sample in the size exclusion column depends on the size of their molecules and diffusion into the pores of the sorbent, as well as on the size of the pores of the stationary phase. In this type of liquid chromatography, the partition coefficient D for the smallest molecules of the analyzed sample, which move in the chromatographic column at the lowest speed, penetrating into the grid of the stationary phase, it is equal to 1, since the mobile phase and the solvent in the pores of the stationary phase have the same composition. In this case, the main equation of column chromatography takes the form Large molecules that do not enter the pores of the stationary phase are eluted from the column along with the mobile phase. For them D= 0, a V R =V m. Such a range of distribution coefficient values (from 0 to 1) is typical only for size exclusion chromatography. All molecules of the analyzed multicomponent substance should be washed out of the column by passing a small volume of solvent from V m before V m +V s and the separation is completed before the solvent peak exits. Therefore, in this type of chromatography, it is necessary to use sufficiently long columns with a large free volume. V m and a large number of pores in the sorbent. The resolution of chromatographic peaks in size exclusion separations can be improved by using gradient elution with mixed solvents. Each sorbent used in size exclusion chromatography is characterized by a certain pore volume and, therefore, has a certain area of separable molecular weights and a certain calibration curve. In this case, the calibration curve characterizing the dependence of the retained volume on the molecular weight or size of the molecules, as a rule, has a complex form. Stationary phases in size exclusion chromatography are selected based on specific analytical tasks. Initially, it is established which solvent system can be used for analysis (aqueous or water-organic). Depending on this, the type of sorbent is determined. If water-soluble samples are to be separated, for example water-swellable cross-linked dextrans (Sephadex) or polyacrylamides (Biogel P) are used as stationary phases. The separation of substances soluble in organic solvents can be carried out on polystyrenes with various degrees of crosslinking, which swell in organic solvents (styrogel, poragel, biobid C). Such swollen gels are generally not pressure stable and allow very low mobile phase flow rates, which increases analysis time. To implement a highly efficient version of size exclusion chromatography, it is necessary to use stationary phases with rigid matrices - silica gels, the disadvantage of which - high adsorption activity - is eliminated by silanization of the surface or selection of an eluent of the appropriate polarity. Substances that can be used as mobile phases in size exclusion chromatography are: completely dissolve the analyzed sample; wet the sorbent well; counteract adsorption of sample components on the sorbent; have low viscosity and toxicity. 1.6.5. Planar chromatography. Planar chromatography includes thin layer and paper chromatography. These types of liquid chromatography are simple in technique, express, do not require expensive equipment, which is their undeniable advantage. The separation of a mixture of substances by these methods can be performed using various chromatographic systems. Therefore, adsorption, distribution, normal and reversed phase, ion-exchange, etc., paper and thin-layer chromatography are distinguished. At present, thin layer chromatography is the most widely used. Paper and thin layer chromatography are similar in technique. Cellulose paper fiber is used as a stationary phase in paper chromatography, in thin-layer chromatography - various sorbents (Al 2 O 3 , silica gel, etc.) deposited in a uniform thin (100-300 μm) layer on a glass, metal or plastic substrate (carrier) . The adsorbent layer on the support may or may not be fixed. Chromatographic separation in planar methods, as well as on a column, is due to the transfer of the components of the analyte by the mobile phase along the layer of the stationary phase at different rates in accordance with the distribution coefficients of the substances to be separated. In both cases, liquid-solid sorbent chromatographic systems (adsorption separation mechanism), liquid-liquid-solid carrier (distribution, ion-exchange and other mechanisms) are used. Various solvents or their mixtures, organic or inorganic acids are used as mobile phases. Practical obtaining planar chromatograms consists in the following. On a strip of chromatographic paper or on a thin layer of sorbent, a starting line is marked with a pencil at a distance of 1 cm from the bottom edge of the strip or plate. A micropipette is used to apply a sample to the start line in the form of a spot with a diameter of not more than 2–3 mm. Then the edge of the strip or plate is lowered into the vessel with the mobile phase, located in a sealed chamber. As the mobile phase rises along the strip or plate and the multiple elementary acts of sorption-desorption, distribution between two liquid phases, ion exchange, etc., which are common in chromatography, occur, the components of the analyzed mixture are separated. The process is usually continued until the solvent has passed from the start line 10 cm. After that, the strip or plate is removed from the chamber and dried. If the components of the analyte are colored, they give the corresponding color spots on the chromatogram. To detect unstained components of the analyte, the chromatogram must be developed. The development of a chromatogram and the detection of sample components can be carried out by various methods and depend on the composition of the analyzed mixtures. The manifestation can be done: -Using UV light. The method is applicable for the detection of substances capable of emitting their own radiation (luminescence) in the visible wavelength range under the action of UV radiation; by means of reagents-developers. For example, the presence of amino acids in the analyzed mixture can be detected using ninhydrin. The dried chromatogram is immersed in a 0.2% solution of ninhydrin in acetone, then dried. The spots corresponding to the various components of the mixture acquire a visual and, as a rule, specific color for each substance; - using iodine. In this case, the detected chromatogram is introduced into a vessel, at the bottom of which there are iodine crystals. Iodine vapors are adsorbed on the spots more strongly, due to which the spots are visualized. Iodine is a non-specific developer reagent. Using specific reagents, it is possible not only to determine the number of components of a mixture, but also to identify the separated substances by the color of the spots. Paper and thin layer chromatography is most often carried out in the so-called ascending variant described above. Quite often, to improve the quality of chromatograms, it is necessary to use more complex variants of planar chromatography, for example, top-down, circular, two-dimensional. In paper or thin layer down chromatography, the analyte is applied to the starting line of the plate or paper strip at the top, and the eluent is fed from the top instead of the bottom. The positive effect of improved separation is due to the contribution of the gravity forces of the components to the separation process. Both upstream and downstream chromatography can be performed in one and two-dimensional versions. In contrast to the one-dimensional flat-bed separation process described above, in two-dimensional chromatographic separation, the separation of the analyzed sample is first carried out in one solvent, then separation is carried out in the direction perpendicular to the first, using another solvent, rotating the first chromatogram by 90 ° C. In circular chromatography, the analyte is applied as a drop in the middle of a plate or sheet of chromatographic paper. One or more solvents are also added dropwise here. This leads to the fact that the resulting chromatogram is a set of radial spots. The position of the spots (zones) that form the separated components of the analyte on a flat chromatogram is characterized by the values of the relative speed of movement of the components in a thin layer R fi. Experimental value R fi defined as the ratio of distance L i, passed i-th component, to the distance L passed by the solvent from the starting line to the front line (Fig. 1.10): Value R fi depends on the nature of the relevant component of the analyzed sample, the nature of the stationary phase, its thickness, the nature and quality of the mobile phase, the method of application of the sample and other factors, but always R fi
1. Value R fi is actually identical to the retention time of a substance or its retention volume, which characterizes the rate of passage of a substance through a chromatographic column, and can be used for qualitative identification of the components of the analyzed sample, and the spot diameter is identical to the height or area of the chromatographic peak and, therefore, to some extent reflects the quantitative content of the substance. Quantitative determination of the composition of the analyzed sample in the simplest case can be assessed visually by the intensity of the intrinsic color of the spots or the intensity of the fluorescent glow of the spots obtained during UV detection. For these purposes, the elution of chromatographic spots is widely used. In this case, the spot obtained on the chromatogram is carefully cut out or scraped off, treated with a suitable solvent, and the resulting solution is examined by the appropriate physicochemical method. You can also use the gravimetric method, in which the corresponding spot is cut out from the chromatogram and weighed. The amount of substance is determined by the difference in the weights of clean paper of the same area and paper with the substance. Paper
(BH
) and thin layer chromatography
(TLC
) according to the separation mechanism belong to partition chromatography
. In the HD method, the carrier is a special chromatographic paper
with certain properties. Stationary phase
is water adsorbed on the surface and pores of paper (up to 20%), mobile - organic solvent, miscible or immiscible with water, water or electrolyte solutions. Mechanism
quite complicated on paper. In the stationary phase, the substance can be retained not only due to dissolution in the water adsorbed by the paper, but also be adsorbed
cellulose directly. Drawn on paper shared components
pass into the mobile phase and move through the capillaries of the paper at different speeds in accordance with interfacial distribution coefficient
each of them. At the beginning chromatography
some of the substance from the paper passes into mobile phase
and move on. When the organic solvent reaches the area of the paper that does not contain the solute, redistribution
: from the organic phase, the substance passes into the aqueous phase, sorbed on paper. Since the components have different affinity for the sorbent
, when moving the eluent, separation occurs: some substances are delayed at the beginning of the path, others move further. Here are combined thermodynamic
(establishment of an equilibrium distribution of substances between phases) and kinetic
(moving components at different speeds) separation aspects. As a result, each component is concentrated on a specific area of the paper sheet: zones of individual components
on the chromatogram
. The use of chromatography on paper has a number of significant disadvantages: the dependence of the separation process on the composition and properties of the paper, the change in the water content in the pores of the paper with changes in storage conditions, a very low chromatography speed (up to several days), and low reproducibility of the results. These shortcomings seriously affect the spread of paper chromatography as a chromatographic method. V TLC method
the process of separating a mixture of substances is carried out in a thin layer sorbent
deposited on an inert solid substrate and provided by the movement mobile phase
(solvent) through the sorbent under the action of capillary forces
.
Byseparation mechanism
distinguish partition, adsorption and ion exchange chromatography
. The separation of the components occurs in these cases either as a result of their different distribution coefficient between the two liquid phases ( partition chromatography
), or due to different adsorbability of compounds by the sorbent ( adsorption chromatography
). The adsorption method is based on different degrees of sorption-desorption of the separated components on the stationary phase. Adsorption
carried out at the expense van der Waals forces
, which is the basis physical adsorption
,
polymolecular
(formation of several adsorbate layers on the surface of the adsorbent) and chemisorption
(chemical interaction of adsorbent and adsorbate). In the case of using such sorbents for TLC as alumina
or silica gel
play a role in separation distribution
, and adsorption
on the developed active surface of the sorbent (150–750 m2/g). Distribution
components of the mixture occurs between water on the surface of the carrier (such adsorbents
, how alumina
,
starch
,
cellulose
,
diatomaceous earth
- and water
form stationary phase
), and the solvent moving through this stationary phase ( mobile phase
). The component of the mixture that is more readily soluble in water moves more slowly than the one that is more readily soluble in the mobile phase. Adsorption
manifested in the fact that between carrier
, for example, aluminum oxide, and the components of the mixture are set adsorption equilibria
- for each component its own, the result of which is different travel speed
mixture components. Two extreme cases can be distinguished: a) the concentration of the substance on the adsorbent is zero. The substance completely dissolves in the mobile phase and is carried away by it (moves along with solvent front
). b) the substance is adsorbed completely, does not interact with the solvent and remains at the start. In practice, with the skillful selection of solvent and adsorbent distribution
compounds are located between these extreme cases, and the substance gradually carried over
from one sorbent layer to another due to simultaneously occurring processes sorption
and desorption
. The solvent passing through the sorbent is called eluent
, the process of moving a substance together with an eluent elution
.
As the liquid moves on the plate, the mixture of substances separates due to the action of forces adsorption
,
distribution
,
ion exchange
or a combination of all of these factors. As a result, separate chromatographic zones
mixture components, i.e. it turns out chromatogram
. Correct selection sorbent
and eluent
determines the efficiency of mixture separation. The mobility of the test substance depends on its affinity for the sorbent and eluting force
(polarity) of the eluent. As the polarity of the compound increases, so does its affinity for the polar sorbent. By increasing the degree of adsorption silica gel
organic compounds are arranged in a row: hydrocarbons<алкилгалогенидыарены<нитросоединения<простые
эфиры <сложные эфиры<альдегиды<спирты<амины<карбоновые
кислоты. В свою очередь for silica gel
eluents can be arranged in ascending order of "polarity" ( eluting power
) and form a series of solvents ( eluotropic series
) in accordance with experimental data: alkanes> benzene> chloroform> diethyl ether> ethyl acetate> alcohols С 2 -С 4> water> acetone> acetic acid> methanol. Thus, a polar compound, alcohol, is adsorbed quite strongly on silica gel and, therefore, moves weakly under the action of such a nonpolar solvent as hexane, and remains near the start line. In turn, the non-polar aromatic hydrocarbon biphenyl is noticeably more mobile in hexane, but even here, to achieve R
f
about 0.5, a more polar aprotic eluent, methylene chloride, is needed. eluent strength
regulate using mixtures of solvents - neighbors in eluotropic series
with different polarity. Currently, TLC mainly uses the following sorbents
: for separation lipophilic substances
silica gel
,
alumina
,
acetylated cellulose
,
polyamides
; to separate hydrophilic substances
cellulose
,
cellulose ion exchangers
,
diatomaceous earth
,
polyamides
. The most important characteristic of a sorbent is its activity
, i.e. ability absorb
(hold) the components of the mixture to be separated. Abroad, a number of firms produce silica gel
,
diatomaceous earth
and alumina
with the addition of 5% gypsum, which is used to fix the sorbent layer in the self-manufacturing of plates. The most common sorbent is silica gel
- hydrated silicic acid, formed by the action of mineral acids on Na 2 SiO 3 and drying of the resulting sol. After grinding the sol, a fraction of a certain grain size is used (indicated on the plate, usually 5-20 microns). silica gel
is an polar sorbent
with OH groups as active sites. It easily sorbs water on the surface and forms hydrogen bonds. Alumina
is a weakly basic adsorbent and is used mainly for the separation of weakly basic and neutral compounds. The disadvantage of plates on aluminum oxide is the obligatory activation of the surface before use in a drying cabinet at a high temperature (100-150 o C) and the low adsorption capacity of the layer compared to silica gel. diatomaceous earth
- adsorbent obtained from natural minerals - diatomaceous earths. The sorbent has hydrophilic properties and a lower adsorption capacity of the layer compared to silica gel. Cellulose:
cellulose-coated thin-layer plates are very effective in separating complex organic molecules. The adsorbent is mainly cellulose balls with a diameter of up to 50 microns, fixed on the carrier with starch. As in paper chromatography, the rise of the solvent front is very slow. Chromatographic analysis
is carried out on industrial plates of Czech production " Silufol
»
(« Silufol
"") of aluminum foil, sometimes reinforced with cardboard, and " Siluplast
» made of plastic coated with a layer of sorbents - silica gel LS 5-40 with starch or gypsum as a binder (up to 10%), or aluminum oxide with or without fluorescent indicators. Records " Silufol
» have a high elution rate, however, they are characterized by low separating power and low sensitivity. During storage, they are sensitive to conditions (humidity, temperature, aggressive media, etc.). Individual firms supply chromatographic plates
with a layer of sorbent of different (usually up to 0.25 mm), but strictly constant thickness (silica gel, cellulose, ion-exchange resin), on glass and substrates made of aluminum foil, plastic, impregnated fiberglass. Plates « Sorbfil
» (TU 26-11-17-89) are produced in Russia on a polymer base (polyethylene terephthalate, grade P) or aluminum substrate (grade AF) with a working layer applied microfractionated silica gel sorbent
grades STX-1A and STX-1VE (produced in the USSR as fractionated silica gel KSKG) with a thickness of 90-120 microns (up to 200 microns), fixed with a special binder - silicasol
. When using silicic acid (silicazole) sol as a binder, which transforms into silica gel after heating, the resulting TLC plates consist of two components: a silica gel layer and a substrate. The thickness uniformity of the sorbent layer on one plate is ±5 µm. Designation example: "Sorbfil-PTSKh-AF-V-UF (10x10)" - high-performance TLC plates on an aluminum substrate, with a phosphor, 10x10 cm. If a glass substrate (grade C) is used, then such plates are reusable and chemically resistant. Their chemical resistance is determined by the chemical resistance of silica gel. As a result, TLC plates can be repeatedly treated with aggressive reagents, for example, with a hot chromium mixture, which removes restrictions on the use of correlating reagents for spot detection and sorbent modification, and allows multiple (up to 30 times or more) regeneration of the plates with a chromium mixture. Glass plates can be cut to the desired size. The mechanical strength of the sorbent layer can be controlled, providing, on the one hand, transportation and multiple processing of the plates and, on the other hand, the possibility of extracting adsorbent layers with separated substances for subsequent washing out of individual compounds from the sorbent and their further study by instrumental methods (IR and UV spectrometry). , X-ray diffraction methods, NMR, etc.). The plates differ in the size of the fractions (particle distribution) of the silica gel that makes up the layer. On analytical plates (grade A) the fraction is 5-17 microns, on highly efficient (grade B) - 8-12 microns. A narrower distribution increases the efficiency of the plates, i.e. the spots of the substances to be separated become more compact (smaller in size) and therefore are better separated when the eluent front passes a shorter distance. On Russian wafers, analytical and high-performance layers do not differ very much, in contrast to wafers from Merck (Germany). High performance plates should be used if substances cannot be separated on analytical plates. Plates of all modifications are produced with a phosphor (UV grade) with 254 nm excitation. The shelf life is not limited, the plates " Sorbfil
» widely tested in the analysis of amino acid derivatives, pesticides, lipids, antibiotics. The TLC method is carried out qualitative identification
components. quantitation
for TLC is also possible, this requires applying the exact amount of substance and additional densitometric studies
with a clear fixation of the intensity of the spots. The most common is semiquantitative method
. It is based on visual comparison
the size and intensity of the spot of a component with the corresponding characteristics of a series of spots of the same substance of different concentrations ( standard reference solutions
). When using a sample in the amount of 1–5 μg, such a simple method provides an accuracy of determining the component content of about 5–10%. Often, in order to determine the components in a sample, it is necessary to carry out sample preparation to obtain a mixture containing the analyzed compounds. Sample preparation is based on the extraction of drugs from the sample with organic solvents ( n-hexane, petroleum ether, diethyl ether, chloroform), purification of the extract and subsequent chromatography in a thin layer of alumina or silica gel. There are several variants of TLC and BC, differing in the way solvent supply
. Depending on the direction of movement of the mobile phase, there are: a)ascending chromatography
the mobile phase is poured onto the bottom of the separation chamber, the paper (plate) is placed vertically; b)descending chromatography
the mobile phase is fed from above and moves down along the sorbent layer of the plate or paper; v)radial chromatography
horizontal advance of the solvent front: the mobile phase is brought to the center of the paper disk (plate), where the mixture to be separated is deposited. The most common is upward elution
(chromatography). Front
eluent
while moving from bottom to top. The choice of solvent (mobile phase) is determined by the nature of the sorbent and the properties of the substances to be separated. Chromatographic separation
BC and TLC methods are carried out in separation chamber
with screwed lid. A quantitative measure of the rate of transfer of a substance using a specific adsorbent and solvent is R value
f
(from English. retention
factor
– delay coefficient, this parameter is analogous to retention time). Position zones of the chromatographed component
set to size coefficient
R
f
equal to the ratio of the velocity of its zone to the velocity of the solvent front. Value R
f
is always less than unity and does not depend on the length of the chromatogram. By the amount R
f
influenced by various factors. So, at low temperatures, substances move more slowly; solvent contamination, adsorbent inhomogeneity, foreign ions in the analyzed solution can change the value R
f
up to 10%. In the selected system, the analytes must have different values R
f
and distributed over the entire length of the chromatogram. It is desirable that the values R
f
lay in the range of 0.05-0.85. In practice, the value R f
calculated as the ratio of the distance l
traveled by the substance to the distance L
passed by the solvent: R
f
=
l/l
(6.1
)
Usually for calculation choose spot center
(Fig. 1). Value R
f
depends on many factors such as chromatographic paper
(its porosity, density, thickness, degree of hydration) and sorbent
(grain size, nature of groups on the surface, layer thickness, its moisture content, nature of the substance, composition of the mobile phase), experimental conditions (temperature, chromatography time, etc.). With the constancy of all chromatography parameters, the value R
f
determined only by the individual properties of each component. Rice. 1. Determination of values on the chromatogram RF
for components A and V, their degree of separation Rs
and the number of theoretical plates N
. The efficiency of BC and TLC also depends on selectivity and sensitivity
reactions used to detect the components of the analyzed mixture. Usually, reagents are used that form colored compounds with the components to be determined - developers. For a more reliable identification of shared components
apply " witnesses
» -solutions standard substances
(in the same solvent as the sample) that are expected to be present in the sample. Standard Substance
applied to the starting line next to the analyzed sample and chromatographed under the same conditions. In practice, a relative value is often used: R
f
rel
=
R
f
x
/
R
f
stand
(6.2)
where R
f
stand
also calculated by formula (6.1). Efficiency
chromatographic separation
characterize number of equivalent theoretical plates
and them tall
. Thus, in the TLC method, the number of equivalent theoretical plates N
A for component A the mixture to be separated is calculated by the formula: N
A
= 16 (l
OA
/
a
(A
))
2
(6.3)
Values l
OA
and a
(A
)
determined as shown in Fig. 6.1. Then the height of the equivalent theoretical plate H
A
is: H
A
=
l
OA
/N
=
a
(A
)
2
/ 16
l
OA
.
(6.4)
Separation is practically possible if R
f
(A)
R
f
(V)
0,1
. To characterize the separation of two components A and V use degree (criterion) of division Rs
: Rs
=
l /
(a
(A)
/
2
+
a
(B)
/
2)=
2
l /
(a
(A)
+
a
(B))
(6.5)
where
l
distance between component spot centers A and V; a
(A)
and a
(V)
spot diameters A and V on the chromatogram (Fig. 6.1). The more Rs
, the more clearly the spots of the components are separated A and V on the chromatogram. Conditions chromatography
are chosen so that the value Rs
different from zero and one, the optimal value Rs
is 0.3
0.7. For rate separation selectivity
two components A and V use separation factor
α
: α
=
l
B
/
l
A
(6.6)
If α = 1, then the components A and V are not separated.